Variational Autoencoders for Molecular Generation: A Comprehensive Guide for Drug Discovery
This article provides a comprehensive exploration of Variational Autoencoders (VAEs) and their transformative role in generative molecular design for drug discovery.
Variational Autoencoders for Molecular Generation: A Comprehensive Guide for Drug Discovery
Abstract
This article provides a comprehensive exploration of Variational Autoencoders (VAEs) and their transformative role in generative molecular design for drug discovery. It covers the foundational principles of VAE architecture, including encoders, decoders, and latent space representation. The content delves into advanced methodological implementations such as graph-based VAEs and transformer-integrated models, alongside their practical applications in de novo drug design. Critical challenges like posterior collapse and molecular representation limitations are addressed with current optimization strategies. The review further examines benchmarking platforms and performance metrics for validating model efficacy, synthesizing key insights to outline future directions for VAE-driven innovation in biomedical research and clinical applications.
Understanding VAEs: From Basic Architecture to Molecular Representation
Variational Autoencoders (VAEs) have emerged as a powerful generative model architecture, finding significant utility in the field of molecular generation research. Since their debut in 2013, VAEs have transformed the landscape of generative modeling by blending deep learning with probabilistic inference [1]. Unlike traditional autoencoders that merely compress and reconstruct data, VAEs learn a continuous, probabilistic latent representation that enables the generation of novel data samples [1] [2]. This capability is particularly valuable in drug discovery, where exploring the vast chemical space of potential drug-like molecules (estimated at 10²³ to 10⁶⁰ compounds) presents a formidable challenge [3]. The core components of a VAE—the encoder, decoder, and latent space—work in concert to enable this generative capability, making them indispensable tools for researchers aiming to design new molecular entities with desired pharmacological properties.
Architectural Fundamentals of VAEs
Core Components and Their Functions
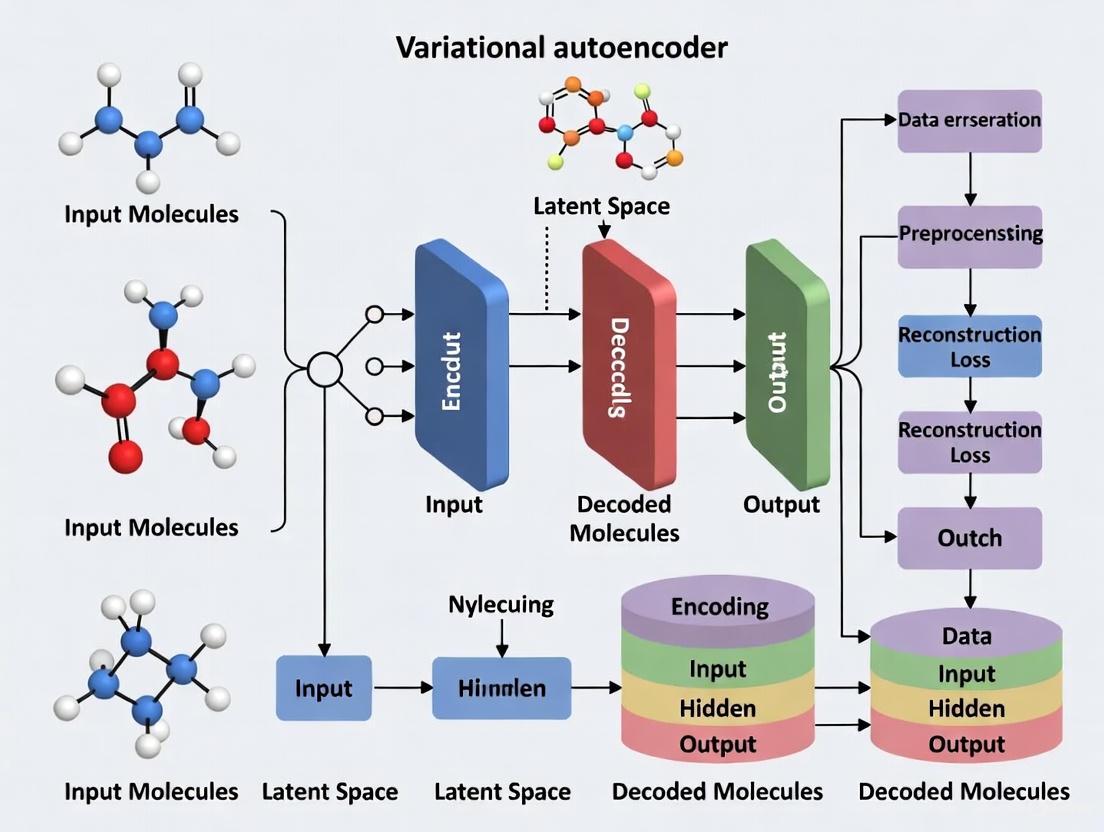
The VAE architecture consists of three primary components working harmoniously: an encoder network, a decoder network, and a structured latent space [1]. Together, these elements form the foundation that enables VAEs to generate new molecular content with remarkable fidelity while ensuring the continuity and completeness of the generated chemical structures.
Table: Core Components of a Variational Autoencoder
| Component | Function | Key Features | Molecular Generation Relevance |
|---|---|---|---|
| Encoder | Maps input data to probabilistic latent representation | Outputs mean (μ) and standard deviation (σ) vectors; uses convolutional or transformer layers | Encodes molecular structures (SMILES, SELFIES, graphs) into continuous latent representations |
| Latent Space | Compressed, probabilistic representation of data | Continuous, structured space following multivariate Gaussian distribution; enables interpolation and sampling | Serves as search space for novel molecules; nearby points decode to structurally similar compounds |
| Decoder | Reconstructs data from latent representations | Uses transposed convolutional or autoregressive layers; outputs reconstructed data samples | Generates novel molecular structures from sampled latent points; ensures syntactic validity |
The Encoder Network
The encoder network in a VAE transforms input data into a latent representation embodying learned attributes [1]. Unlike traditional autoencoders, VAE encoders produce probabilistic representations by outputting both mean and standard deviation vectors for each dimension of the latent space [2]. During this process, input data (x) maps to latent variables (z), commonly written as z|x [1]. A critical sampling layer acts as a constraint point, enabling latent representations that facilitate both reconstruction and generation.
In molecular generation applications, encoder designs have evolved from simple convolutional networks to sophisticated architectures like Transformers and Graph Neural Networks (GNNs) to better handle molecular representations [4] [5]. For instance, the Transformer Graph VAE (TGVAE) employs a molecular graph as input data, capturing complex structural relationships within molecules more effectively than string-based models [4].
The Latent Space Representation
In VAEs, the latent space provides a continuous, probabilistic condensed representation of input data [1]. Each attribute is represented probabilistically, with latent vector values associated with comparable reconstructions from input data [1]. This statistical distribution, determined by mean and variance parameters, ensures minor latent space changes generate consistent new data points [1] [6].
The latent space must exhibit two critical types of regularity: continuity (nearby points yield similar content when decoded) and completeness (any point sampled yields meaningful content) [6]. These properties are enforced by structuring the latent space to follow a known distribution, typically a standard Gaussian, through the Kullback-Leibler (KL) divergence component of the loss function [1] [6]. For molecular generation, this structured latent space enables smooth interpolation between molecular structures and provides a foundation for optimizing molecular properties [3] [5].
The Decoder Network
The decoder network restores the original input using encoded latent variables, essentially reversing the encoding process [1]. Its goal is learning a transformation that takes latent space variables (z) and maps them back into data (x) closely approximating initial inputs [1]. Decoder output dimensions typically match input data dimensions, enabling it to function as a generative model producing new examples similar to training data.
In advanced molecular VAEs, decoder architectures have progressed from recurrent neural networks to autoregressive Transformer decoders, which generate molecular sequences token by token while maintaining chemical validity [5]. For example, STAR-VAE employs an autoregressive Transformer decoder trained on SELFIES representations to guarantee 100% syntactic validity of generated molecules [5].
Mathematical Foundations
VAEs rely on solid mathematical principles for both functionality and efficiency. A well-organized continuous latent space forms their core, critical for enhanced generative capabilities [1]. The key mathematical concepts include variational inference, KL divergence, and the Evidence Lower Bound (ELBO) [1].
The VAE objective function combines reconstruction loss with KL divergence:
Where the first term represents reconstruction error and the second term regularizes the latent space by minimizing the divergence between the learned distribution q(z|x) and the prior distribution p(z) [2]. The full loss function can be written as:
This formulation is known as the Evidence Lower Bound (ELBO), which balances high-quality data reconstruction with appropriate regularization of the latent space [1]. The reparameterization trick enables efficient training by expressing the random latent variable z as a deterministic function of the encoder parameters and an independent random variable: z = μ + σ ⊙ ε, where ε ∼ N(0,1) [1] [7].
Experimental Protocols for Molecular VAE Implementation
Protocol 1: Building a Basic Molecular VAE
Objective: Implement a fundamental VAE for molecular generation using SMILES or SELFIES representations.
Materials and Software:
- Python 3.8+
- Deep learning framework (TensorFlow/Keras or PyTorch)
- RDKit for chemical validation
- MOSES benchmark tools for evaluation
Procedure:
- Data Preprocessing:
- Curate molecular dataset (e.g., from PubChem, ZINC)
- Convert structures to SELFIES representation to guarantee syntactic validity [5]
- Tokenize sequences and create vocabulary
- Split data into training/validation sets (80/20 ratio)
Encoder Implementation:
- Implement embedding layer for token inputs
- Design encoder architecture (e.g., Transformer with 4 layers, 8 attention heads)
- Add output layers for mean (μ) and log-variance (log σ²) of latent distribution
- Implement sampling layer using reparameterization trick
Decoder Implementation:
- Implement autoregressive Transformer decoder with masked self-attention
- Add output projection layer to vocabulary size
- Apply softmax activation for token probability distribution
Training Configuration:
- Initialize optimizer (Adam with learning rate 0.0001)
- Set batch size to 128 and train for 100 epochs
- Use categorical cross-entropy for reconstruction loss
- Weight KL divergence term with annealing schedule (β from 0.01 to 1.0)
Validation:
- Monitor reconstruction accuracy and validity rate
- Evaluate generated molecules using MOSES benchmark metrics [3]
- Assess latent space organization using dimensionality reduction (t-SNE, UMAP)
Protocol 2: Conditional Molecular Generation
Objective: Extend VAE for property-guided molecular generation.
Procedure:
- Property Prediction Module:
- Add property prediction head to encoder output
- Pre-train on labeled molecular property data (e.g., solubility, toxicity)
- Use multi-task learning during VAE training
Conditional Training:
- Concatenate property vectors with latent representations
- Modify decoder to accept conditional inputs
- Implement classifier-guided sampling for generation
Evaluation:
- Assess property optimization capabilities using GuacaMol benchmark [5]
- Evaluate target-specific generation using docking scores
Diagram Title: VAE Architecture for Molecular Generation
Advanced Applications in Molecular Research
Addressing Posterior Collapse in Molecular VAEs
Posterior collapse remains a significant challenge in molecular VAEs, where the model fails to utilize the latent space effectively, limiting the diversity of generated molecules [3]. The PCF-VAE approach addresses this by reparameterizing the loss function and incorporating a diversity layer between the latent space and decoder [3]. This architecture modification, combined with GenSMILES representations that simplify molecular complexity, has demonstrated validity rates of 95-98% across different diversity levels while maintaining 100% uniqueness in generated structures [3].
Table: Performance Comparison of Advanced Molecular VAEs
| Model | Architecture | Representation | Validity Rate | Uniqueness | Novelty | Internal Diversity |
|---|---|---|---|---|---|---|
| PCF-VAE [3] | VAE with diversity layer | GenSMILES | 95.01-98.01% | 100% | 93.77-95.01% | 85.87-89.01% |
| STAR-VAE [5] | Transformer VAE | SELFIES | High (MOSES benchmark) | Competitive | Competitive | Structured latent space |
| TGVAE [4] | Transformer-Graph VAE | Molecular graph | Enhanced vs. string models | Improved diversity | Novel structures | Effective structural capture |
Latent Space Visualization and Analysis
Visualizing the VAE latent space helps understand how the model discerns and assimilates fundamental data structure [8]. By condensing input data into compact latent space, VAEs extract pivotal attributes while neglecting superfluous details [8]. Methods like t-SNE and PCA reduce dimensions, enabling understanding of learned features through visible clusters and patterns [8] [7].
In ensemble visualization applications, VAEs transform spatial features of ensembles into latent spaces following multivariate standard Gaussian distributions, enabling analytical computation of confidence intervals and density estimation [8]. This capability is valuable for understanding uncertainty in molecular property predictions and exploring chemical space neighborhoods around promising candidate molecules.
Diagram Title: Molecular VAE Training and Optimization Workflow
Research Reagent Solutions
Table: Essential Tools for Molecular VAE Research
| Reagent/Tool | Function | Application Example |
|---|---|---|
| SELFIES Representation [5] | Guarantees 100% syntactically valid molecular strings | STAR-VAE uses SELFIES to ensure validity in generated molecules |
| Graph Neural Networks [4] | Processes molecular graph structures directly | TGVAE employs GNNs to capture structural relationships in molecules |
| Low-Rank Adaptation (LoRA) [5] | Enables parameter-efficient finetuning with limited data | STAR-VAE uses LoRA for fast adaptation with property data |
| GenSMILES [3] | Simplified SMILES representation reducing complexity | PCF-VAE uses GenSMILES to enhance robustness and diversity |
| Transformer Architectures [5] | Handles long-range dependencies in molecular sequences | Replaces RNNs in modern VAEs for improved sequence modeling |
| Molecular Property Predictors [5] | Provides conditioning signals for guided generation | Integrated into conditional VAE frameworks for target-oriented design |
Future Directions and Challenges
The field of molecular VAEs continues to evolve with several promising research directions. Hybrid models that combine VAEs with other generative approaches such as GANs or diffusion models show potential for enhancing sample quality and diversity [1] [9]. Addressing the challenge of posterior collapse remains an active area of investigation, with approaches like PCF-VAE demonstrating significant improvements in generating diverse, valid molecules [3].
Future work may focus on better integration of 3D structural information, improved conditioning mechanisms for multi-property optimization, and more efficient training strategies for scaling to larger chemical spaces [9] [5]. As molecular VAEs mature, they are poised to become increasingly valuable tools in the drug discovery pipeline, enabling more efficient exploration of chemical space and acceleration of therapeutic development.
Variational inference provides a scalable framework for approximate probabilistic inference, which has become a cornerstone of modern machine learning applications, including the generation of molecular structures for drug discovery. The fundamental challenge that necessitates variational inference is the intractability of the posterior distribution in complex latent variable models. When working with latent variable models, we often have observed variables (e.g., molecular structures) and latent variables (e.g., hidden representations capturing chemical properties). In a Bayesian framework, we specify a prior over the latent variables 𝑝(𝐳) and a likelihood function 𝑝(𝐱|𝐳) that connects latents to observables. The cornerstone of Bayesian inference is the posterior distribution 𝑝(𝐳|𝐱) = 𝑝(𝐱,𝐳)/𝑝(𝐱), which requires computation of the marginal likelihood or evidence 𝑝(𝐱) = ∫ 𝑝(𝐱,𝐳) 𝑑𝐳. This integral is generally intractable for complex models, as it involves integration over all possible configurations of latent variables, often with exponential computational cost [10].
Key Mathematical Concepts
Kullback-Leibler Divergence
The Kullback-Leibler (KL) divergence measures the similarity between two probability distributions. Given the true posterior 𝑝(𝐳|𝐱) and a variational approximation 𝑞(𝐳), the KL divergence is defined as:
KL ( 𝑞(𝐳) ‖ 𝑝(𝐳|𝐱) ) = ∫ 𝑞(𝐳) log [ 𝑞(𝐳) / 𝑝(𝐳|𝐱) ] 𝑑𝐳 = - ∫ 𝑞(𝐳) log [ 𝑝(𝐳|𝐱) / 𝑞(𝐳) ] 𝑑𝐳
This divergence is non-negative (KL ≥ 0) and zero only when 𝑞(𝐳) equals 𝑝(𝐳|𝐱). However, it is not symmetric (KL(𝑝‖𝑞) ≠ KL(𝑞‖𝑝)) and does not satisfy the triangle inequality, thus not a true distance metric. The forward KL (𝑝‖𝑞) tends to be "mode-covering" (averaging), while the reverse KL (𝑞‖𝑝) tends to be "mode-fitting" [10].
The Evidence Lower Bound (ELBO)
Direct minimization of KL(𝑞(𝐳)‖𝑝(𝐳|𝐱)) is intractable because it requires the very evidence term we cannot compute. The derivation of the Evidence Lower Bound (ELBO) provides a solution through mathematical transformation:
KL ( 𝑞(𝐳) ‖ 𝑝(𝐳|𝐱) ) = 𝔼₍𝐳∼𝑞₎ [ log 𝑞(𝐳) ] - 𝔼₍𝐳∼𝑞₎ [ log 𝑝(𝐳|𝐱) ] = 𝔼₍𝐳∼𝑞₎ [ log 𝑞(𝐳) ] - 𝔼₍𝐳∼𝑞₎ [ log 𝑝(𝐱,𝐳) - log 𝑝(𝐱) ] = 𝔼₍𝐳∼𝑞₎ [ log 𝑞(𝐳) - log 𝑝(𝐱,𝐳) ] + log 𝑝(𝐱)
Since KL divergence is non-negative, we have: log 𝑝(𝐱) ≥ 𝔼₍𝐳∼𝑞₎ [ log 𝑝(𝐱|𝐳) ] - KL( 𝑞(𝐳) ‖ 𝑝(𝐳) )
The right-hand side is the ELBO. Maximizing the ELBO minimizes the KL divergence and provides a lower bound to the log evidence [10].
Visualizing the Core Relationships
The following diagram illustrates the fundamental relationship between evidence, KL divergence, and ELBO:
Application to Molecular Generation
Variational Autoencoders for Molecular Design
In molecular generation, variational autoencoders (VAEs) leverage the variational inference framework to learn continuous latent representations of molecular structures. The encoder network approximates the posterior 𝑞(𝐳|𝐱), while the decoder network parameterizes the likelihood 𝑝(𝐱|𝐳). During training, the ELBO objective is maximized, forcing the model to learn chemically meaningful representations while regularizing the latent space [3].
Current research addresses specific challenges in molecular VAEs, particularly posterior collapse, where the model fails to utilize the latent space effectively, resulting in low diversity of generated molecules. The PCF-VAE approach introduces reparameterization of the loss function and transforms SMILES strings into GenSMILES to reduce complexity and enhance robustness [3].
Advanced Variational Methods
Recent advancements incorporate more expressive latent distributions. The Variational Mean Flow (VMF) framework models the latent space as a mixture of Gaussians rather than a unimodal Gaussian, better capturing the multimodal nature of molecular distributions. This approach enables efficient one-step inference while maintaining generation quality and diversity [11].
Another innovation combines variational inference with causal modeling through Causality-Aware Transformers (CAT), which enforce directional dependencies in molecular assembly through masked attention mechanisms, ensuring causally coherent generation of molecular substructures [11].
Experimental Protocols & Implementation
Protocol: Implementing a Basic Molecular VAE
Purpose: To create a variational autoencoder for generating novel molecular structures with desired properties.
Materials:
- Molecular dataset (e.g., ZINC, QM9)
- Graph neural network libraries (PyTorch Geometric, DGL)
- Chemical validation tools (RDKit, OpenBabel)
Procedure:
- Data Preparation:
- Convert molecular structures to appropriate representation (SMILES, graphs)
- Apply standardization: neutralize charges, remove solvents, normalize tautomers
- Split dataset into training/validation/test sets (80/10/10)
Model Architecture:
- Encoder: Graph isomorphism network (GIN) or graph attention network (GAT)
- Latent space: Multivariate Gaussian (mean and variance vectors)
- Decoder: Recurrent network (for SMILES) or graph generation network
Training Configuration:
- Objective: ELBO = 𝔼[log p(𝑥|𝑧)] - β⋅KL(𝑞(𝑧|𝑥)‖𝑝(𝑧))
- Optimizer: Adam (learning rate: 0.001, β₁: 0.9, β₂: 0.999)
- Batch size: 128-256 depending on memory constraints
- β annealing: Gradually increase β from 0 to 1 over training
Validation:
- Chemical validity: Percentage of valid molecular structures
- Uniqueness: Fraction of duplicate molecules in generated set
- Novelty: Percentage of generated molecules not in training set
- Diversity: Structural diversity metrics (Tanimoto similarity, scaffold diversity)
Protocol: Mitigating Posterior Collapse in Molecular VAEs
Purpose: To address the posterior collapse problem where the model ignores latent codes.
Procedure:
- Architectural Modifications:
- Use weaker decoders (e.g., single-layer LSTM instead of multi-layer)
- Add skip connections from encoder to decoder
- Implement stochastic recurrent connections in decoder
Training Techniques:
- Apply KL annealing: gradually increase weight of KL term in ELBO
- Use free bits: enforce minimum KL per latent dimension
- Add auxiliary losses: property prediction, fragment conservation
Alternative Objectives:
- Implement InfoVAE: Add mutual information term to objective
- Use β-VAE: Weight KL term more heavily (β > 1)
Workflow for Molecular Generation with VAEs
The complete experimental workflow for molecular generation integrates each component of the variational framework:
Research Reagent Solutions
Table 1: Essential computational tools for molecular generation with variational autoencoders
| Reagent/Tool | Function | Application Notes |
|---|---|---|
| RDKit | Cheminformatics toolkit for molecule manipulation and validation | Essential for processing SMILES, calculating molecular properties, and validating generated structures [3] |
| PyTorch Geometric | Graph neural network library | Implements graph encoders for molecular structures; supports message passing and graph pooling [4] |
| TensorFlow Probability | Probabilistic programming | Provides distributions, bijectors, and probabilistic layers for building VAE components |
| MOSES Benchmark | Evaluation framework for molecular generation | Standardized metrics for validity, uniqueness, novelty, and diversity [3] |
| Graphviz | Graph visualization | Creates publication-quality diagrams of molecular structures and model architectures |
Performance Metrics and Comparative Analysis
Table 2: Performance comparison of VAE-based molecular generation methods
| Model | Validity (%) | Uniqueness (%) | Novelty (%) | Internal Diversity | Key Innovation |
|---|---|---|---|---|---|
| PCF-VAE [3] | 95.01-98.01 | 100 | 93.77-95.01 | 85.87-89.01% | Posterior collapse mitigation via loss reparameterization |
| TGVAE [4] | High (exact values not reported) | High | High | High | Transformer + GNN integration for graph-based generation |
| MolSnap [11] | 100 | Not reported | Up to 74.5 | Up to 70.3% | Variational Mean Flow with mixture priors |
| Standard VAE [3] | Typically <90% | Variable | Variable | Often limited | Baseline for comparison |
Advanced Applications and Future Directions
The integration of variational inference with molecular generation continues to evolve. Recent approaches combine VAEs with flow-based methods, where normalizing flows transform simple distributions into complex ones through a series of invertible transformations, providing more flexible posterior approximations [11].
In genome-wide association studies (GWAS), variational inference enables scalable analysis of large biobanks. The Quickdraws method uses stochastic variational inference with spike-and-slab priors to increase association power without sacrificing computational efficiency [12].
For single-cell RNA sequencing data, probabilistic matrix factorization with variational inference (PMF-GRN) infers gene regulatory networks by decomposing gene expression into latent factors representing transcription factor activity and regulatory relationships [13].
These applications demonstrate how the mathematical foundations of variational inference—KL divergence and ELBO—enable scalable, probabilistic modeling across diverse scientific domains, particularly in molecular generation where handling complexity and uncertainty is paramount for effective drug discovery.
The reparameterization trick is a foundational technique in machine learning that enables the training of probabilistic models, most notably Variational Autoencoders (VAEs), through standard gradient-based methods. In the context of molecule generation for drug discovery, this trick is indispensable. It allows researchers to efficiently explore vast chemical spaces by learning smooth, continuous latent representations of molecular structures. By providing a pathway for gradient flow through random sampling nodes, the reparameterization trick facilitates the optimization of complex objective functions that balance molecular validity, diversity, and desired pharmacological properties. This document details the application of this technique and its associated sampling processes within molecular generative models, providing structured protocols and data for research and development professionals.
Technical Foundation of the Reparameterization Trick
The Problem: Non-Differentiable Sampling
In a standard VAE, the encoder neural network does not output a deterministic latent vector z. Instead, it learns the parameters (mean μ and standard deviation σ) of a Gaussian distribution, from which the latent vector z is sampled. This sampling operation is inherently stochastic and non-differentiable, which blocks the flow of gradients during backpropagation, preventing the model from learning the parameters μ and σ [14] [15].
The core objective is to optimize the Evidence Lower Bound (ELBO), which includes a reconstruction loss and a regularization term (Kullback-Leibler divergence). Computing the gradient of the expectation term, ∇φ E_{z∼qφ(z|x)}[f(z)], with respect to the distribution parameters φ is not straightforward due to the sampling step [16].
The Solution: Reparameterization
The reparameterization trick addresses this by decoupling the randomness from the learnable parameters. Instead of sampling z directly from N(μ, σ²), the random variable z is expressed as a deterministic function of the parameters and an independent noise variable. For a Gaussian distribution, this is achieved as follows:
Deterministic Function: z = gφ(ε, x) = μφ(x) + σφ(x) ⊙ ε
Noise Variable: ε ∼ N(0, I)
Here, ε is an auxiliary noise variable sampled from a standard normal distribution, μφ(x) and σφ(x) are the outputs of the encoder network, and ⊙ denotes element-wise multiplication [14] [16]. This reformulation moves all stochasticity to the variable ε, which is independent of the parameters φ. The path from the parameters φ to the latent variable z is now entirely deterministic and differentiable, allowing gradients to flow through the model via z to the encoder, enabling end-to-end training with stochastic gradient descent [15] [16].
Table 1: Reparameterization Formulations for Common Distributions
| Distribution | Reparameterization Function z = gφ(ε) |
Noise Distribution p(ε) |
|---|---|---|
| Gaussian | z = μ + σ ⊙ ε |
ε ∼ N(0, I) |
| Exponential | z = -log(ε) / λ |
ε ∼ Uniform(0, 1) |
Application in Molecular Variational Autoencoders
In molecular generation, VAEs map input molecular representations (e.g., SMILES, SELFIES) into a structured latent space. The reparameterization trick is the engine that makes this learning process feasible.
Molecular VAE Architecture and Workflow
The following diagram illustrates the flow of data and gradients in a molecular VAE that utilizes the reparameterization trick.
Diagram 1: Molecular VAE with Reparameterization. This workflow shows how gradients flow from the reconstruction loss back through the deterministic latent vector z to the encoder's parameters.
Quantitative Performance in Molecular Generation
The effectiveness of VAEs trained with the reparameterization trick is measured by their ability to generate valid, unique, and novel molecules. The following table summarizes key performance metrics from recent state-of-the-art molecular VAE models on standard benchmarks.
Table 2: Performance Metrics of Molecular VAEs on Generation Tasks
| Model | Validity (%) | Uniqueness (%) | Novelty (%) | Internal Diversity (IntDiv %)* | Key Feature |
|---|---|---|---|---|---|
| PCF-VAE [3] | 95.01 - 98.01 | 100 | 93.77 - 95.01 | 85.87 - 89.01 | Mitigates posterior collapse |
| STAR-VAE [5] | High (SELFIES) | - | - | - | Transformer-based, uses SELFIES |
| VAE-CYC [17] | Good | - | - | Good | Cyclical annealing to prevent collapse |
| MolMIM [17] | High | - | - | Good | Alternative architecture |
Internal Diversity (IntDiv) measures the structural variety within a set of generated molecules. A higher value indicates greater diversity [3].
Experimental Protocols
Protocol: Implementing the Reparameterization Trick for a Gaussian VAE
This protocol details the steps to implement the reparameterization trick in a molecular VAE using a Gaussian latent space.
Objective: To enable gradient-based optimization of a VAE for molecular generation by implementing the reparameterization trick.
Materials:
- Hardware: GPU-enabled workstation.
- Software: Python 3.x, PyTorch or TensorFlow, RDKit, molecular dataset (e.g., ZINC, ChEMBL).
Procedure:
- Encoder Forward Pass:
- Pass an input molecule (as a SMILES or SELFIES string, suitably encoded) through the encoder network.
- The encoder outputs two vectors: the mean
μand the logarithm of the variancelog σ². Usinglog σ²ensures the standard deviation is always positive during optimization [15].
Sampling with Reparameterization:
Decoder Forward Pass & Loss Computation:
- Pass the latent vector
zthrough the decoder network to reconstruct the original input molecule. - Compute the total loss
L, which is the negative ELBO:L = L_reconstruction + β * D_KL(q_φ(z|x) || p(z))where:
- Pass the latent vector
Backward Pass & Optimization:
- Compute the gradients of the total loss
Lwith respect to all model parameters (includingφfrom the encoder) using backpropagation. Becausezis a deterministic function ofφ, gradients can flow through it. - Update the model parameters using a stochastic gradient descent optimizer (e.g., Adam).
- Compute the gradients of the total loss
Troubleshooting:
- Posterior Collapse: If the KL divergence term collapses to zero early in training, the encoder ignores the input. Mitigation strategies include KL annealing (gradually increasing
βfrom 0) or using a cyclical annealing schedule [17] [3]. - Low Validity: When using SMILES strings, invalid outputs are common. Consider switching to SELFIES representations, which guarantee 100% syntactic validity [5].
Protocol: Optimizing Molecules in Latent Space with Reinforcement Learning
This protocol describes a method for optimizing generated molecules for specific properties by combining a pre-trained VAE with a latent space optimization algorithm.
Objective: To generate molecules with optimized properties (e.g., high drug-target affinity, specific lipophilicity) by navigating the continuous latent space of a pre-trained VAE.
Materials:
- A pre-trained molecular VAE with a well-structured latent space.
- A reward function
R(m)that scores a moleculembased on the desired properties.
Procedure:
- Latent Space Continuity Check:
- Before optimization, verify the continuity of the VAE's latent space. Encode a set of test molecules to their latent points
z₀. - Perturb
z₀by adding Gaussian noise with varying variancesσto createz' = z₀ + ε, ε ∼ N(0, σI). - Decode the perturbed points and measure the Tanimoto similarity between the original and perturbed molecules. A gradual decrease in similarity with increasing
σindicates a continuous space suitable for optimization [17].
- Before optimization, verify the continuity of the VAE's latent space. Encode a set of test molecules to their latent points
Optimization Loop:
- Initialization: Start from an initial latent point
z(e.g., encoding of a seed molecule or a random point). - Action: At each step
t, the optimization algorithm (e.g., Proximal Policy Optimization - PPO) proposes a stepΔzin the latent space, moving to a new pointz_t = z_{t-1} + Δz. - Evaluation: Decode
z_tto a moleculem_tand compute the rewardR(m_t). - Feedback: The reward signal
R(m_t)is used to update the policy of the optimization algorithm, encouraging it to explore regions of latent space that decode to high-scoring molecules. - Iteration: Repeat steps b-d for a fixed number of iterations or until convergence [17].
- Initialization: Start from an initial latent point
Candidate Selection:
- Decode the final latent points from the optimization trajectory to obtain the optimized candidate molecules.
- Validate the chemical structures and properties using external tools (e.g., RDKit, molecular docking simulations).
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for Molecular VAE Research
| Item Name | Function / Purpose | Example Use Case |
|---|---|---|
| ZINC Database | A public repository of commercially available chemical compounds for training and benchmarking. | Serves as the primary dataset for training generative models [17]. |
| ChEMBL Database | A large-scale bioactivity database for drug discovery. | Used for training property prediction models and conditional generation [18]. |
| SELFIES | A 100% robust molecular string representation. | Replaces SMILES in VAEs to guarantee generation of syntactically valid molecules [5]. |
| RDKit | Open-source cheminformatics software. | Used for parsing SMILES/SELFIES, calculating molecular properties, and validating generated structures [17]. |
| PCF-VAE Loss | A modified VAE loss function designed to prevent posterior collapse. | Improves the diversity and validity of generated molecules in de novo drug design [3]. |
| Low-Rank Adapters (LoRA) | A parameter-efficient fine-tuning method. | Adapts a large pre-trained VAE to new property prediction tasks with limited labeled data [5]. |
This application note provides a detailed comparison of SMILES strings and molecular graphs as molecular representations, contextualized within Variational Autoencoder (VAE)-based molecular generation research. We include structured data, experimental protocols, and visualization tools to aid researchers in selecting and implementing these representations for drug discovery applications.
Molecular representation is a foundational step in computational chemistry, bridging the gap between chemical structures and their biological properties [19]. In VAE-based molecular generation, the choice of representation directly influences the model's ability to learn a continuous, meaningful latent space from which valid and novel molecules can be decoded [20] [21]. SMILES and molecular graphs are two dominant representations, each with distinct strengths and limitations for deep learning applications. This note provides a practical framework for their evaluation and use.
Core Representation Formats: A Technical Comparison
SMILES (Simplified Molecular Input Line Entry System) is a line notation that uses short ASCII strings to describe molecular structure [22] [23]. Molecular Graphs represent a molecule as a set of nodes (atoms) and edges (bonds), directly encoding its topological structure [20].
Table 1: Quantitative Comparison of Representation Performance in VAE Models
| Feature | SMILES-Based VAE (e.g., CVAE, GVAE) | Graph-Based VAE (e.g., JT-VAE, NP-VAE) |
|---|---|---|
| Representation Type | Sequential, string-based [19] | Topological, graph-based [20] |
| Inherent Validity of Generated Structures | Low; many outputs are invalid SMILES [20] [21] | High; outputs are inherently valid molecular graphs [20] |
| Handling of Large/Complex Molecules | Limited; struggles with complex structures like large natural products [20] | Excellent; newer models (e.g., NP-VAE) are designed for large compounds [20] |
| Inclusion of Stereochemistry | Supported in isomeric SMILES [22] [24] | Can be incorporated as node/edge parameters [20] |
| Example Reconstruction Accuracy | Lower than graph-based models [20] | NP-VAE demonstrated higher reconstruction accuracy [20] |
Table 2: Qualitative Analysis of Strengths and Weaknesses
| Aspect | SMILES Strings | Molecular Graphs |
|---|---|---|
| Primary Strengths | Compact, human-readable, vast existing support in cheminformatics [22] [24] | Natural representation of structure, high validity rates, flexible feature attachment [20] |
| Key Limitations | Non-uniqueness, sensitivity to small syntax errors, abstract representation [20] [25] | Computational complexity, requires specialized canonicalization, historically limited to smaller molecules [20] |
| Best-Suited VAE Tasks | Initial prototyping, exploration of chemical language models [19] | Generation of syntactically valid, complex molecules, and scaffold hopping [20] [19] |
Experimental Protocols for VAE Modeling
Protocol: Building a SMILES-Based VAE (CVAE)
This protocol outlines the steps for constructing a SMILES-based Conditional VAE (CVAE) for multi-property molecular generation [21].
1. Data Preprocessing and Canonicalization
- Input: Raw molecular dataset (e.g., from ZINC database [21]).
- Procedure:
- Canonicalization: Use a cheminformatics toolkit (e.g., RDKit) to convert all structures into a unique, canonical SMILES string. This ensures consistent representation [21].
- Tokenization: Treat the SMILES string as a sequence of characters. Pad the end of the string with a unique termination character (e.g., 'E').
- Vectorization: Convert each character into a one-hot encoded vector.
- Property Conditioning: Create a condition vector (c) containing normalized values of target molecular properties (e.g., Molecular Weight, LogP, TPSA). For integer properties like HBD/HBA, use one-hot encoding [21].
2. Model Architecture and Training
- Encoder: A Recurrent Neural Network (RNN), typically with LSTM cells, processes the one-hot encoded SMILES sequence and the condition vector to produce a latent vector (z) [21].
- Decoder: Another RNN (the decoder) takes the latent vector (z) and the same condition vector (c) to reconstruct the SMILES string sequence step-by-step [21].
- Training Objective: Minimize the loss function of the CVAE:
E[logP(X|z,c)] - D_KL[Q(z|X,c) || P(z|c)], where the first term is the reconstruction loss and the second is the Kullback-Leibler divergence, which regularizes the latent space [21].
3. Molecular Generation and Validation
- Sampling: Generate molecules by sampling a latent vector (z) from the prior distribution
N(0, I)and concatenating it with a desired condition vector (c). - Decoding: Use a "stochastic write-out" process where each character in the SMILES string is sampled from the probability distribution output by the decoder's softmax layer. Perform multiple decodings (e.g., 100x) per latent vector to maximize valid output [21].
- Validation: Pass the generated SMILES string to RDKit to check for chemical validity and calculate its properties for comparison with the target condition [21].
Protocol: Building a Graph-Based VAE (NP-VAE)
This protocol is based on the NP-VAE model, which is designed to handle large molecular structures with 3D complexity [20].
1. Molecular Graph Decomposition and Featurization
- Input: Large, complex molecular structures (e.g., natural products from DrugBank).
- Procedure:
- Graph Construction: Represent the molecule as a hydrogen-suppressed molecular graph where nodes are atoms and edges are bonds [26] [20].
- Tree Decomposition: Use a graph decomposition algorithm (e.g., Junction Tree algorithm) to break the molecular graph into meaningful chemical substructures or fragments, which are organized into a tree structure [20].
- Feature Extraction: Encode each node (atom) in the graph using features such as chemical element, Morgan vertex degree, and electronic structure (e.g., 1s2, 2p3). For isomeric SMILES, chirality and bond stereochemistry are also encoded [26] [20].
2. Model Architecture and Latent Space Construction
- Encoder: A Graph Neural Network (GNN) or Tree-LSTM processes the molecular graph (or its junction tree) to map it into a latent vector (z) [20].
- Decoder: The decoder network reconstructs the molecular graph from the latent vector, typically by assembling the predicted substructures. This ensures the output is always a valid molecular graph [20].
- Latent Space: The model constructs a continuous, low-dimensional latent space. Exploration of this space allows for the generation of novel compound structures optimized for specific functions [20].
3. Generation and Functional Optimization
- Exploration: Sample points from the latent space or interpolate between known active molecules to generate novel molecular structures.
- Validation: The graph-based decoding process guarantees 100% syntactically valid molecules. The validity of the chemical structure itself is checked with RDKit [20].
- Docking Analysis: Generated molecules with desired properties can be virtually screened using molecular docking simulations to assess their potential as drug candidates [20].
The Scientist's Toolkit: Essential Research Reagents & Software
Table 3: Key Software and Resources for Molecular VAE Research
| Tool Name | Type | Primary Function in VAE Research |
|---|---|---|
| RDKit [20] [21] | Cheminformatics Library | Checks SMILES validity, calculates molecular properties, handles file format conversion. |
| CORAL [26] | QSAR Software | Calculates optimal descriptors from SMILES and molecular graphs for model building. |
| ZINC Database [21] | Molecular Library | Provides large, publicly available datasets of drug-like molecules for model training. |
| DrugBank [20] | Pharmaceutical Database | Source of approved drug and natural product structures for training complex generative models. |
| NP-VAE [20] | Deep Learning Model | Specialized graph-based VAE for handling large natural product structures with chirality. |
| JT-VAE [20] | Deep Learning Model | A foundational graph-based VAE that uses junction tree decomposition for high reconstruction accuracy. |
The field of molecular representation is dynamically evolving. While graph-based VAEs currently show superior performance in generating valid and complex molecules, SMILES-based models remain relevant for specific applications and as a component of chemical language models [20] [19]. Future directions point toward hybrid models and the increased use of multimodal learning and contrastive learning frameworks to create even more powerful and interpretable chemical latent spaces, further accelerating AI-driven drug discovery [19].
The Role of Latent Space in Capturing Chemical Similarity
In the field of molecular generation research, the latent space of a Variational Autoencoder (VAE) serves as a crucial low-dimensional mathematical representation that captures the essential features of chemical compounds [20]. This continuous, probabilistic space is fundamental for enabling tasks such as molecule generation, optimization, and the meaningful exploration of chemical properties [1] [5]. By learning to project high-dimensional, complex molecular structures into a structured, lower-dimensional manifold, the VAE's latent space provides a powerful framework for navigating the vast chemical universe and identifying novel compounds with desired characteristics [20] [27]. Its ability to implicitly encode chemical similarity—where molecules with similar structures or properties are located near each other in the latent space—makes it an indispensable tool for modern computational drug discovery [5] [28].
Latent Space Fundamentals in Molecular VAEs
The latent space in a VAE is a compressed, probabilistic representation of input data, learned by aligning the distribution of encoded data points with a prior distribution, typically a unit Gaussian [1] [27]. This is achieved through the optimization of the Evidence Lower Bound (ELBO), which balances two objectives: reconstruction loss, ensuring the decoded output closely matches the original input, and the Kullback-Leibler (KL) divergence, which regularizes the structure of the latent space to be continuous and smooth [1]. This structured continuity is what allows for meaningful interpolation and navigation within the latent space, as small changes in the latent vector correspond to coherent and gradual changes in the generated molecular structure [20] [27].
Unlike traditional autoencoders that may learn a non-smooth, disjointed latent manifold, the variational formulation enforces a well-behaved space [27]. This property is critical for molecular optimization, as it allows for the use of efficient continuous optimization techniques, such as Bayesian optimization, to traverse the latent space and discover molecules with optimized properties [28]. The latent space thus acts as a "chemical cartography" tool, mapping discrete molecular structures onto a continuous domain where their relationships can be quantified and exploited for generative design [20].
Quantitative Performance of Molecular VAE Frameworks
Different VAE architectures have been developed to efficiently capture chemical similarity and generate valid molecules. The table below summarizes the performance of several key models on standard benchmarks, highlighting their effectiveness in reconstruction and generation.
Table 1: Performance Comparison of Molecular VAE Frameworks
| Model Name | Key Architecture | Molecular Representation | Reconstruction Accuracy | Validity | Key Innovation |
|---|---|---|---|---|---|
| NP-VAE [20] | Graph-based VAE with Tree-LSTM | Molecular graph | ~90% (on evaluation dataset) | 100% (fragment-based) | Handles large, complex molecules & chirality |
| STAR-VAE [5] | Transformer Encoder-Decoder | SELFIES | Matches/exceeds baselines on GuacaMol & MOSES | High (SELFIES guarantee) | Scalable pretraining & property-guided generation |
| JT-VAE [20] | Junction Tree Graph VAE | Molecular graph | High (for small molecules) | High | Treats molecular graphs as tree structures |
| CLaSMO [28] | Conditional VAE (CVAE) | Molecular graph | N/A (Modification-based) | N/A | Scaffold optimization via latent space Bayesian optimization |
| TGVAE [4] | Transformer & Graph Neural Network | Molecular graph | Outperforms existing approaches | High & Diverse | Combins Transformer, GNN, and VAE |
A critical challenge in molecular VAEs is the choice of representation. Early models like CVAE used SMILES strings, but often suffered from low validity, as many generated strings did not correspond to valid molecules [20] [5]. Subsequent innovations have largely shifted to graph-based representations (e.g., JT-VAE, NP-VAE) or modern string-based representations like SELFIES, which guarantee 100% syntactic validity and thus improve the utility of the latent space for reliable generation [5] [4].
Experimental Protocols for Latent Space Application
Protocol: Constructing a Chemical Latent Space with NP-VAE
This protocol details the procedure for building a latent space for large, complex molecules, such as natural products, using the NP-VAE model [20].
Data Curation and Preparation
- Source: Obtain molecular structures from databases like DrugBank and natural product libraries [20].
- Preprocessing: Standardize structures and compute molecular features/fingerprints (e.g., ECFP). For large molecules, apply the model's decomposition algorithm to break compounds into fragment units and convert them into tree structures [20].
- Split: Divide the dataset into training, validation, and test sets (e.g., 76,000/5,000/5,000 compounds) [20].
Model Training and Latent Space Formation
- Architecture: Configure the NP-VAE encoder and decoder using Tree-LSTM networks to process the molecular tree structures [20].
- Training Loop: Train the model by minimizing the VAE loss function (ELBO), which combines reconstruction loss and KL divergence. Use the validation set for early stopping [20].
- Encoding: Pass the training and test sets through the trained encoder to project each molecule into a point in the low-dimensional latent space, defined by the mean (μ) and standard deviation (σ) vectors [20].
Latent Space Evaluation
- Reconstruction Accuracy: For each test compound, perform multiple stochastic encodings and decodings (e.g., 10x10). Calculate the proportion where the output structure exactly matches the input [20].
- Generalization Assessment: Sample latent vectors from the prior distribution ( N(0, I) ), decode them, and use RDKit to check the validity of the generated structures [20].
Protocol: Property-Guided Molecular Generation with STAR-VAE
This protocol outlines the use of a transformer-based VAE for generating molecules conditioned on specific properties [5].
Large-Scale Pretraining
- Data: Curate a large dataset of drug-like molecules (e.g., ~79 million from PubChem). Apply standard drug-likeness filters (e.g., Molecular Weight ≤ 600) [5].
- Representation: Convert all molecules to SELFIES representations to ensure syntactic validity. Tokenize the SELFIES strings [5].
- Model Pretraining: Train the STAR-VAE model (Transformer encoder and autoregressive Transformer decoder) on the SELFIES corpus using the ELBO objective. This creates a general-purpose chemical latent space [5].
Conditional Finetuning
- Property Predictor: Attach a property prediction head to the pretrained encoder. Finetune it on a smaller dataset labeled with the target property (e.g., docking scores) [5].
- Conditional Generation Signal: The property predictor supplies a conditioning signal that is consistently applied to the latent prior, the inference network, and the decoder during generation [5].
- Parameter-Efficient Adaptation: Employ Low-Rank Adaptation (LoRA) in both the encoder and decoder to adapt the model to the conditional generation task with limited labeled data [5].
Evaluation of Conditional Generation
- Benchmarking: Evaluate the unconditional generation performance on benchmarks like GuacaMol and MOSES for validity, uniqueness, and diversity [5].
- Property-Specific Assessment: For conditional generation (e.g., optimizing docking scores), generate molecules and compare the distribution of their target properties against a baseline VAE to demonstrate a statistically significant shift towards improved values [5].
Protocol: Scaffold-Based Optimization with CLaSMO
This protocol describes a sample-efficient method for optimizing existing molecular scaffolds by performing Bayesian optimization in the latent space of a Conditional VAE [28].
Data Preparation for Scaffold Modification
- Input: Define a set of molecular scaffolds to be optimized.
- Substructure Enumeration: Create a dataset of valid substructures and their corresponding attachment points. For each scaffold, generate examples of how substructures can be bonded to it [28].
- Conditioning Feature Extraction: For each attachment point on a scaffold, compute the atomic environment features that will serve as the conditioning input for the CVAE [28].
Training the Conditional VAE
- Model: Train a CVAE where the encoder learns a latent distribution for a substructure, and the decoder generates a substructure conditioned on the scaffold's atomic environment features [28].
- Objective: The model learns to generate chemically compatible substructures that can be integrated into the scaffold.
Latent Space Bayesian Optimization (LSBO)
- Define Objective: Formulate the target property (e.g., binding affinity, solubility) as a black-box function to be maximized.
- Optimization Loop: a. Select a scaffold and an attachment point to modify. b. Use the CVAE encoder to project known substructures for that context into the latent space. c. Fit a Bayesian optimization surrogate model (e.g., Gaussian Process) to the latent points and their property values. d. Propose the next latent point to evaluate by maximizing an acquisition function (e.g., Expected Improvement). e. Decode the proposed latent point into a new substructure, attach it to the scaffold, and evaluate the property of the new molecule (or use a predictor). f. Update the surrogate model with the new data point and repeat [28].
- Constrained Output: The process maintains a similarity constraint to the original scaffold, ensuring synthesizability.
Essential Research Reagent Solutions
The following table catalogs key computational tools and resources essential for conducting research on latent space and chemical similarity.
Table 2: Key Research Reagents and Tools for Molecular VAE Research
| Reagent / Resource | Type | Function in Research | Example Use Case |
|---|---|---|---|
| RDKit [20] | Cheminformatics Software | Handles molecular I/O, fingerprint calculation, and validity checks. | Evaluating the chemical validity of molecules generated from the latent space. |
| SELFIES [5] | Molecular Representation | String-based representation guaranteeing 100% syntactic validity. | Used in STAR-VAE to prevent generation of invalid molecular strings. |
| ECFP [20] | Molecular Fingerprint | Represents molecular structure as a bit vector; used as input feature. | Providing structural features for the VAE encoder to learn meaningful representations. |
| PubChem [5] | Chemical Database | Large-scale source of drug-like molecules for model training. | Curating a dataset of ~79 million molecules for pretraining STAR-VAE. |
| GuacaMol / MOSES [5] | Benchmarking Framework | Standardized benchmarks for evaluating generative model performance. | Quantifying the validity, uniqueness, and diversity of molecules generated by a trained VAE. |
| Low-Rank Adaptation (LoRA) [5] | Fine-tuning Technique | Efficiently adapts large pre-trained models to new tasks with limited data. | Fine-tuning STAR-VAE for property-guided generation without full retraining. |
| Bayesian Optimization [28] | Optimization Algorithm | Efficiently optimizes expensive black-box functions in continuous spaces. | Navigating the latent space of CLaSMO to find molecules with optimal properties. |
Workflow Visualization
The following diagram illustrates the high-level logical workflow of a VAE for molecular generation, from input to generation and optimization, integrating the components discussed in the protocols.
Diagram 1: Molecular VAE Workflow. This figure outlines the core process of molecular encoding, latent space formation, and molecule generation/optimization, highlighting the pathways for both reconstruction and conditional generation. BO: Bayesian Optimization.
Advanced VAE Architectures and Their Application in De Novo Drug Design
Variational Autoencoders (VAEs) have emerged as a powerful deep learning framework for generative tasks, particularly in domains with complex, structured data like chemistry and drug discovery. Among these, graph-based VAEs represent a significant advancement as they operate directly on molecular graphs, inherently preserving the structural relationships between atoms. This application note details the operational principles, performance metrics, and experimental protocols for two prominent graph-based VAE architectures—the Junction Tree VAE (JT-VAE) and the Natural Product-oriented VAE (NP-VAE)—within the context of molecule generation research. We focus on their enhanced ability to generate chemically valid and structurally accurate molecular structures compared to earlier methods.
Junction Tree VAE (JT-VAE)
The JT-VAE addresses the critical challenge of molecular graph reconstruction by decomposing a molecule into a hierarchical, tree-like structure of chemical substructures, or "junction trees." This decomposition constrains the generation process to chemically plausible steps, dramatically improving the validity of the output.
- Core Principle: The model encodes a molecule using two parallel encoders: one for the original molecular graph and another for its junction tree. The junction tree represents the molecule as a tree where nodes are chemically valid fragments (e.g., rings, functional groups) and edges represent their adjacency. This simplifies the complex graph into a tractable tree structure.
- Architecture and Training: The model uses a Graph Neural Network (GNN) to encode the molecular graph and a Tree-based GNN to encode the junction tree. These two latent representations are concatenated to form the final molecular embedding. The decoder first generates a junction tree and then assembles the final molecular graph by combining the generated fragments, guided by the molecular graph embedding [29]. Training involves an initial deterministic autoencoder phase, followed by fine-tuning with a Kullback-Leibler (KL) divergence penalty to regularize the latent space [29].
Natural Product-Oriented VAE (NP-VAE)
The NP-VAE was developed to handle large, complex molecular structures that are intractable for earlier models, such as natural products with significant 3D complexity and chirality.
- Core Principle: NP-VAE is a graph-based VAE that combines an algorithm for decomposing compound structures into fragment units and converting them into tree structures with Extended Connectivity Fingerprints (ECFP) and a Tree-LSTM network [30].
- Advancements over Predecessors: NP-VAE represents a significant improvement over models like JT-VAE and HierVAE, with 12 million parameters. Its key innovations include the ability to handle chirality (an essential factor for 3D structure and biological activity) and a mechanism to train the chemical latent space by incorporating functional information alongside structural data [30]. This allows for the generation of novel compounds optimized for a target property.
Performance and Comparative Analysis
The performance of graph-based VAEs is quantitatively assessed based on their ability to accurately reconstruct input molecules (reconstruction accuracy) and to generate novel, valid, and unique molecular structures.
Table 1: Comparative Performance of Generative Models on Molecular Tasks
| Model | Reconstruction Accuracy | Validity | Key Strengths and Applicable Scope |
|---|---|---|---|
| JT-VAE [29] | ~76% (on QM9 dataset with HOMO prediction task) | High (by design) | High validity for small molecules; enables property prediction and optimization via latent space. |
| NP-VAE [30] | >80% (outperforms baselines on St. John's dataset) | 100% (generates in substructure units) | Handles large, complex structures & chirality; suited for natural product-like compounds. |
| CVAE (SMILES-based) [30] | Lower than graph-based models | Very Low (majority invalid) | Pioneering application of VAE; now largely superseded by graph-based methods. |
| HierVAE [30] | Lower than NP-VAE | High | Handles larger compounds with repeating structures; cannot consider stereochemistry. |
The data from Table 1 demonstrates the clear superiority of graph-based models over SMILES-based approaches in generating chemically valid structures. NP-VAE shows a marked improvement in reconstruction accuracy, establishing it as a high-performance generative model for complex molecular structures [30].
Table 2: Latent Space Utilization for Molecular Optimization (e.g., HOMO energy)
| Model / Strategy | Property Prediction Performance (e.g., HOMO) | Successful Optimization Capability |
|---|---|---|
| JT-VAE with Regression Model [29] | Achieved state-of-the-art results in HOMO prediction. | Yes: Latent space allows for gradient-based search to find molecules with a predefined HOMO value. |
| NP-VAE with Functional Latent Space [30] | Latent space trained with functional information. | Yes: Enables generation of novel compounds optimized for a target function by exploring the latent space. |
Experimental Protocols
Protocol 1: Training a JT-VAE for Property Prediction and Optimization
This protocol outlines the steps for pre-training a JT-VAE and employing its latent space for molecular property optimization [29].
Model Pre-training
- Dataset: Utilize a large-scale molecular dataset such as ZINC for initial training.
- Two-Phase Training:
- Phase 1 (Deterministic AE): Train the encoder-decoder pair as a standard autoencoder to minimize reconstruction error.
- Phase 2 (VAE Tuning): Introduce the KL divergence penalty between the latent vectors and a standard normal distribution to regularize the latent space.
- Objective: Learn a robust latent space that captures the fundamental distribution of molecular structures.
Regression Model Training
- Dataset: Use a property-specific dataset like QM9, which includes quantum chemical properties such as HOMO energy.
- Procedure: With the pre-trained JT-VAE encoder frozen, train a feedforward neural network (FFNN) regressor. The regressor maps latent vectors (
Z) from the JT-VAE to the target property values (e.g., HOMO). - Architecture: A typical regressor can consist of two hidden layers (e.g., size 1024) with ReLU activation functions [29].
Molecular Optimization (Reverse-QSAR)
- Input: A target property value
v₀(e.g., a specific HOMO energy). - Optimization Loop:
- Initialize a latent vector
Z(e.g., from a known molecule's encoding). - Define the loss
L = |v₀ - f_R(Z)|, wheref_Ris the trained regressor. - Use gradient descent within the latent space to minimize
Lby updatingZ, keeping the weights of the encoder and regressor frozen. - Stop when
f_R(Z)is sufficiently close tov₀.
- Initialize a latent vector
- Decoding: Pass the optimized latent vector
Zthrough the JT-VAE decoder to generate the molecular structureD(Z)with the desired property [29].
- Input: A target property value
Protocol 2: Evaluating Model Generalization Ability
This protocol describes a standard evaluation method for assessing the reconstruction and generative capabilities of a molecular VAE [30].
- Data Splitting: Divide a standardized dataset (e.g., St. John et al.'s dataset with 76,000 training, 5,000 validation, and 5,000 test compounds) into training, validation, and test sets.
- Model Training: Train the VAE model on the training set.
- Reconstruction Accuracy (Generalization Ability):
- For each test compound, perform multiple stochastic encodings and decodings (e.g., 10 encodings, each decoded 10 times, for 100 total outputs per test compound).
- Calculate the proportion of output structures that exactly match the input test compound.
- Validity and Novelty:
- Sample a large number of latent vectors (e.g., 1000) from the prior distribution
N(0, I). - Decode each vector multiple times.
- Use a toolkit like RDKit to determine the proportion of outputs that are chemically valid molecules.
- Check the uniqueness of the generated valid molecules against the training set.
- Sample a large number of latent vectors (e.g., 1000) from the prior distribution
Visualization of Workflows
JT-VAE Encoding and Optimization Pathway
NP-VAE Molecular Decomposition Workflow
The Scientist's Toolkit: Research Reagents and Solutions
Table 3: Essential Computational Tools for Graph-Based Molecular VAEs
| Item / Resource | Function / Description | Example Tools / Libraries |
|---|---|---|
| Molecular Datasets | Provide structured data for training and benchmarking models. | ZINC database (for general molecules); QM9 (with quantum properties); DrugBank & Natural Product libraries [30]. |
| Cheminformatics Toolkit | Handles molecular I/O, validity checks, fingerprint generation, and stereochemistry. | RDKit [30]. |
| Deep Learning Framework | Provides flexible environment for building and training complex neural networks. | PyTorch, TensorFlow. |
| Graph Neural Network Library | Offers pre-built modules for implementing graph convolutions and message passing. | PyTorch Geometric (PyG), Deep Graph Library (DGL). |
| Latent Space Analysis Package | Aids in visualization and interpolation within the learned latent space. | scikit-learn (for PCA, t-SNE). |
The exploration of chemical space for novel drug candidates represents a monumental challenge in pharmaceutical research, necessitating advanced computational approaches for efficient molecular design. Within the framework of variational autoencoder (VAE) research for molecule generation, two prominent strategies have emerged for processing the Simplified Molecular Input Line-Entry System (SMILES) representation: Grammar Variational Autoencoders (GVAEs) and Character-Level Recurrent Neural Networks (Char-RNNs). These approaches fundamentally differ in how they interpret and generate SMILES strings, with GVAEs employing grammatical constraints to ensure syntactic validity and Char-RNNs utilizing statistical sequence modeling at the character level. This article provides detailed application notes and experimental protocols for these methodologies, enabling researchers to effectively implement and evaluate these models for de novo molecular design tasks. The structured comparison and standardized protocols presented herein aim to facilitate reproducibility and advance the field of AI-driven drug discovery.
Theoretical Foundations
SMILES Representation for Molecules
The Simplified Molecular Input Line-Entry System (SMILES) provides a string-based representation that encodes molecular structures as linear sequences of characters, offering a compact and human-readable format for computational processing [31]. This notation utilizes an alphabet of characters where elemental symbols (e.g., 'C' for carbon, 'N' for nitrogen) are combined with special characters representing chemical features: '-' for single bonds, '=' for double bonds, '#' for triple bonds, and numerals to indicate ring closures [32] [33]. For example, benzene is represented in aromatic SMILES notation as "c1ccccc1" [32]. Despite its widespread adoption, standard SMILES notation suffers from limitations including limited token diversity, lack of chemical information within individual tokens, and potential for generating invalid structures due to its context-free nature [31] [20].
Grammar-Based VAEs for Molecular Design
Grammar Variational Autoencoders (GVAEs) represent a significant advancement over character-level models by incorporating formal grammatical constraints to ensure syntactic validity of generated outputs [34]. The fundamental innovation of GVAEs lies in their treatment of structured discrete data whose validity can be characterized by a formal grammar. Rather than processing raw SMILES characters, GVAEs encode molecules as sequences of production rules derived from context-free grammars (CFGs) or molecular hypergraph grammars (MHGs) [34]. This approach guarantees that all decoder outputs comply with the grammatical rules of SMILES syntax, effectively eliminating invalid structure generation.
The GVAE framework employs a standard VAE architecture where the encoder receives a sequence of grammar production rules representing the parse of an input molecule. These production sequences are typically one-hot encoded into binary matrices and processed through deep convolutional neural networks or recursive LSTM architectures to output parameters (μ, σ²) of a Gaussian variational posterior [34]. The decoder maps latent codes to valid production rule sequences using a recurrent neural network (LSTM or GRU) with dynamic masking that ensures only syntactically valid derivations can be produced at each decoding step [34].
For molecular applications specifically, molecular hypergraph grammars (MHGs) have been developed to overcome the limitations of context-free grammars in expressing chemical constraints such as atom valency [34]. MHGs generalize CFGs by representing molecules as hypergraphs, with productions that operate on hyperedges and rigorously adhere to molecular validity constraints including regularity and cardinality [34].
Character-Level RNNs for Molecular Generation
Character-Level Recurrent Neural Networks (Char-RNNs) approach molecular generation as a sequence modeling problem, analogous to statistical language modeling in natural language processing [32] [33]. These models learn the probability distribution of the next character in a SMILES string given a sequence of previous characters, enabling the generation of novel molecules one character at a time [32]. Char-RNNs operate directly on the character-level representation of SMILES strings without explicit grammatical constraints, relying instead on the statistical patterns learned from large datasets of valid molecules.
The architecture typically employs Long Short-Term Memory (LSTM) networks, which are well-suited for capturing long-range dependencies in sequential data [32] [35]. The model processes input sequences through an embedding layer, followed by multiple LSTM layers that maintain hidden states to capture contextual information, and finally a fully-connected output layer that predicts the probability distribution over the next possible character [33]. During training, the model learns to maximize the likelihood of the training sequences, effectively capturing the statistical regularities of valid SMILES strings in its parameters.
Performance Comparison and Quantitative Assessment
Table 1: Comparative Performance Metrics of SMILES-Based Generative Models
| Model | Validity Rate | Uniqueness | Novelty | Reconstruction Accuracy | Internal Diversity (intDiv) |
|---|---|---|---|---|---|
| GVAE | 99% [34] | 100% [34] | 93.77% [3] | 53.7% [34] | 85.87-89.01% [3] |
| MHG-VAE | 100% [34] | 100% [34] | 94.71% [3] | 94.8% [34] | 85.87-86.33% [3] |
| Char-RNN | ~90% [20] | >95% [32] | ~85% [32] | N/A | ~80% [32] |
| PCF-VAE | 95.01-98.01% [3] | 100% [3] | 93.77-95.01% [3] | >90% [3] | 85.87-89.01% [3] |
Table 2: Optimization Performance for Molecular Properties
| Model | Best Penalized logP | Synthesizability Improvement | Binding Affinity Improvement | Drug-likeness (QED) |
|---|---|---|---|---|
| GVAE | -9.57 [34] | +5% [31] | +6% [31] | 0.7 [20] |
| MHG-VAE | 5.24 [34] | +6% [31] | +7% [31] | 0.72 [20] |
| Char-RNN | 2.91 [20] | +3% [31] | +4% [31] | 0.68 [20] |
| PCF-VAE | 4.85 [3] | +5% [3] | +6% [3] | 0.71 [3] |
Experimental Protocols
Protocol 1: Implementing Grammar VAE for Molecular Generation
Objective: To implement and train a Grammar VAE model for generating valid molecular structures with optimized properties.
Materials:
- ZINC or ChEMBL dataset (SMILES representations)
- RDKit or OpenBabel cheminformatics toolkit
- Python 3.7+ with PyTorch/TensorFlow
- GPU-enabled computational environment
Procedure:
Data Preprocessing:
- Curate a dataset of drug-like molecules in SMILES format (e.g., ZINC database containing ~250,000 compounds)
- Apply standardization of SMILES representation using RDKit's CanonicalSMILES
- Implement grammar derivation from SMILES strings using context-free grammar rules
- Split data into training (80%), validation (10%), and test sets (10%)
Model Architecture Configuration:
- Implement encoder network with 3 convolutional layers (filter sizes: 9, 9, 10; stride: 3, 3, 2)
- Design decoder with stacked LSTM layers (2 layers, 512 hidden units)
- Set latent space dimension to 196 continuous variables
- Implement rule masking mechanism during decoding to ensure grammatical validity
Training Protocol:
- Initialize model weights using Xavier initialization
- Set batch size to 128 and initial learning rate to 0.001
- Use Adam optimizer with β1=0.9, β2=0.999
- Implement learning rate reduction on plateau (factor=0.5, patience=5 epochs)
- Train for maximum 100 epochs with early stopping (patience=10 epochs)
Validation and Testing:
- Evaluate reconstruction accuracy on test set
- Assess validity of generated molecules using RDKit's SMILES parser
- Calculate novelty (% of generated molecules not in training set)
- Measure uniqueness (% of unique molecules among generated set)
Diagram 1: GVAE Architecture for Molecular Generation
Protocol 2: Character-Level RNN for SMILES Generation
Objective: To train a character-level RNN model for generating novel molecular structures using SMILES notation.
Materials:
- ChEMBL database (≥2 million bio-active molecules) [32]
- PyTorch deep learning framework
- NVIDIA GPU with ≥8GB memory
- Custom Python scripts for data processing
Procedure:
Data Preparation:
- Extract SMILES strings from ChEMBL database
- Create character vocabulary from all unique characters in SMILES
- Implement character-to-integer mapping dictionary
- Convert all SMILES to one-hot encoded representations
- Generate training sequences with fixed length (100 characters)
Model Architecture:
- Implement character embedding layer (dimensionality: 256)
- Design multi-layer LSTM network (2 layers, 512 hidden units each)
- Add dropout regularization (rate=0.2) between LSTM layers
- Implement fully-connected output layer with softmax activation
Training Configuration:
- Set batch size to 128 and sequence length to 100
- Use cross-entropy loss function and Adam optimizer
- Implement gradient clipping (max norm: 5.0)
- Train for 50 epochs with teacher forcing ratio of 0.5
Sampling and Generation:
- Implement priming with starting characters (e.g., 'C' or 'c')
- Use temperature-based sampling for diversity control
- Generate molecules of varying lengths (50-120 characters)
- Validate generated SMILES using RDKit chemical validation
Diagram 2: Char-RNN Architecture for SMILES Generation
Protocol 3: Latent Space Optimization for Property Enhancement
Objective: To optimize molecular properties through latent space exploration of trained VAEs.
Materials:
- Pre-trained GVAE or Char-RNN model
- Bayesian optimization library (e.g., GPyOpt)
- Molecular property predictors (e.g., for logP, QED, synthesizability)
- Target protein structure for binding affinity calculations
Procedure:
Latent Space Characterization:
- Encode training set molecules to latent representations
- Perform principal component analysis to identify major variation axes
- Train property predictors on latent representations (Random Forest or MLP)
Bayesian Optimization Setup:
- Define objective function combining multiple properties
- Set acquisition function (Expected Improvement)
- Initialize with 100 random points in latent space
- Run optimization for 500 iterations
Multi-objective Optimization:
- Balance conflicting properties (e.g., potency vs. solubility)
- Implement Pareto front identification
- Generate diverse candidate molecules from optimal latent points
Validation:
- Synthesize top candidates (10-20 molecules)
- Experimental testing of key properties
- Iterative model refinement based on experimental results
Advanced Hybridization Techniques
Recent advancements in SMILES representation have led to hybrid approaches that enhance model performance. The SMI+AIS(N) representation method seamlessly integrates standard SMILES tokens with Atom-In-SMILES (AIS) tokens, which incorporate local chemical environment information into a single token [31]. This hybrid approach maintains SMILES simplicity while enriching the representation with critical chemical context, addressing the token frequency imbalance inherent in standard SMILES [31].
The SMI+AIS representation demonstrates significant improvements in binding affinity (7% improvement) and synthesizability (6% increase) compared to standard SMILES in molecular generation tasks [31]. This enhancement stems from the method's ability to differentiate chemical elements based on their chemical context without introducing unnecessary tokens for less frequent elements [31]. The hybridization effectively mitigates the token frequency imbalance by replacing frequently observed SMILES tokens (e.g., 'C') with multiple AIS tokens distinguished by chemical environment (e.g., '[cH;R;CC]', '[c;R;CCC]', and '[CH3;!R;C]') [31].
Table 3: Research Reagents and Computational Tools
| Resource | Type | Application | Access |
|---|---|---|---|
| ZINC Database | Compound Library | Training data for generative models | Public |
| ChEMBL Database | Bioactive Molecules | Training data for drug-like molecules | Public |
| RDKit | Cheminformatics | SMILES validation and manipulation | Open Source |
| PyTorch/TensorFlow | Deep Learning Frameworks | Model implementation | Open Source |
| MOSES Benchmark | Evaluation Platform | Standardized model assessment | Public |
| OpenBabel | Chemical Toolbox | Format conversion and descriptor calculation | Open Source |
Grammar VAEs and Character-Level RNNs represent complementary approaches to SMILES-based molecular generation, each with distinct advantages and limitations. GVAEs provide guaranteed syntactic validity through grammatical constraints and demonstrate superior performance in reconstruction accuracy and latent space organization, making them ideal for targeted molecular optimization [34]. Char-RNNs offer flexibility and have demonstrated remarkable success in generating novel molecular structures with properties correlating well with those of the training molecules [32] [33]. The emerging hybrid approaches, such as SMI+AIS representation, further enhance model performance by incorporating chemical context directly into the molecular representation [31]. As the field advances, the integration of these methodologies with experimental validation cycles will play a crucial role in accelerating drug discovery and development pipelines.
The exploration of chemical space, estimated to contain approximately 10^60 possible small molecules, represents a monumental challenge in modern drug discovery [20]. Structural diversity within compound libraries is crucial for discovering new pharmaceutical compounds, particularly those derived from natural products which often exhibit complex structures and high biological activity [20]. Variational Autoencoders (VAEs) have emerged as powerful deep learning frameworks for constructing chemical latent spaces—projections of molecular structures into mathematical space based on molecular features [20] [36]. However, existing molecular VAEs have struggled with large, complex molecular structures with 3D complexity, such as natural products with essential chiral centers [20] [36].
The Natural Product-oriented Variational Autoencoder (NP-VAE) addresses these limitations as a specialized deep learning method capable of handling hard-to-analyze datasets and large molecular structures with stereochemical complexity [20] [37]. By effectively constructing chemical latent spaces that include natural compounds, NP-VAE enables comprehensive library analysis and generation of novel compound structures with optimized functions, representing a significant advancement in computational drug discovery [20] [36].
NP-VAE Architecture and Technical Innovations
Core Architectural Components
NP-VAE incorporates several technical innovations that enable its handling of complex molecular structures:
Graph-Based Molecular Representation: Unlike SMILES-based approaches that struggle with validity issues, NP-VAE represents compounds as graph structures defined by adjacency relationships between atoms, ensuring chemically valid output generation [20].
Tree-Structured Decomposition: The model employs an algorithm for effectively decomposing compound structures into fragment units and converting them into tree structures, facilitating handling of large molecular architectures [20].
Chirality Handling: A crucial innovation of NP-VAE is its ability to manage stereochemistry, an essential factor in the 3D complexity of compounds, particularly relevant for natural products [20] [38]. The model incorporates chirality information through Extended Connectivity Fingerprints (ECFP) [38].
Tree-LSTM Integration: NP-VAE utilizes Tree-LSTM, a specialized recurrent neural network, to process the tree-structured molecular representations, enabling effective handling of hierarchical molecular patterns [20] [38].
Workflow and Latent Space Construction
The NP-VAE workflow transforms complex molecular structures into a continuous latent space representation that captures essential structural and functional features. The encoder component processes input molecular structures through graph decomposition and Tree-LSTM networks to generate latent variables, while the decoder reconstructs molecular structures from these latent representations [20]. This continuous latent space enables exploration of structural diversity and generation of novel compounds through interpolation and optimization within the learned space.
Performance Evaluation and Comparative Analysis
Reconstruction Accuracy and Generalization
NP-VAE demonstrates superior performance in reconstruction accuracy and generalization capability compared to existing state-of-the-art models. Using the standardized evaluation dataset from St. John et al.'s study (divided into 76,000 training compounds, 5,000 validation compounds, and 5,000 test compounds), NP-VAE achieved higher reconstruction accuracy for test compounds than all baseline models [20]. The reconstruction accuracy was evaluated using the Monte Carlo method, where for each test compound, 10 encodings were performed with 10 decodings each, resulting in 100 output compounds per test compound [20].
Table 1: Comparative Performance of Molecular Generative Models
| Model | Reconstruction Accuracy | Validity | Handling of Large Compounds | Chirality Support |
|---|---|---|---|---|
| NP-VAE | Highest | 100% (fragment-based generation) | Excellent | Yes |
| HierVAE | Moderate | High | Good | No |
| JT-VAE | High | High | Limited | No |
| CG-VAE | Moderate | High | Limited | No |
| CVAE | Low | Low (requires validation) | Limited | No |
| MoFlow (Flow-based) | 100% (theoretical) | High | Limited | No |
NP-VAE's fragment-based generation approach ensures 100% validity of output compounds, addressing a significant limitation of SMILES-based methods that often generate invalid chemical representations [20]. This makes NP-VAE particularly suitable for generating complex natural product-like structures that adhere to chemical rules.
Application in Electrolyte Additive Design
Beyond drug discovery, NP-VAE has demonstrated exceptional performance in material science applications. In electrolyte additive design for lithium-ion batteries, a fine-tuned NP-VAE model generated approximately 1,000 novel candidate molecules and predicted their HOMO and LUMO values with remarkable accuracy [38]. When validated against Density Functional Theory (DFT) calculations, the model achieved exceptionally low mean absolute errors of 0.04996 eV for HOMO and 0.06895 eV for LUMO predictions, demonstrating its capability for accurate electrochemical property prediction [38].
Experimental Protocols
Protocol 1: Latent Space Construction and Compound Reconstruction
Purpose: To construct a chemical latent space from complex molecular structures and evaluate reconstruction accuracy [20].
Materials:
- Compound libraries (e.g., DrugBank, natural product databases)
- RDKit cheminformatics toolkit [20]
- NP-VAE implementation (Tree-LSTM encoder, MLP decoder) [20] [38]
- Training dataset: 76,000 compounds [20]
- Validation dataset: 5,000 compounds [20]
- Test dataset: 5,000 compounds [20]
Procedure:
- Data Preparation:
- Curate compound structures from source databases
- Convert structures to graph representations with chirality information
- Apply train/validation/test split (76,000/5,000/5,000 compounds)
Model Training:
- Initialize NP-VAE with Tree-LSTM encoder and MLP decoder
- Configure training parameters: batch size, learning rate, latent dimension
- Train model to minimize reconstruction loss and KL divergence
- Validate reconstruction accuracy using Monte Carlo method [20]
Reconstruction Evaluation:
- For each test compound: perform 10 encodings → 10 decodings each (100 total outputs per compound)
- Calculate proportion of exactly matched input-output structures
- Assess chemical validity of all outputs using RDKit [20]
Latent Space Analysis:
- Project compounds into latent space using encoder
- Analyze clustering patterns and interpolation continuity
- Identify regions corresponding to structural features
Troubleshooting Tips:
- For poor reconstruction: Increase latent dimension size or Tree-LSTM hidden units
- For invalid structures: Verify proper chirality handling in ECFP encoding
- For training instability: Adjust KL divergence weight in loss function
Protocol 2: Property-Guided Molecular Generation
Purpose: To generate novel compound structures with optimized target properties through latent space exploration [20] [38].
Materials:
- Pretrained NP-VAE model
- Property prediction model (HOMO/LUMO, bioactivity, etc.)
- Optimization algorithm (gradient descent, Bayesian optimization)
- DFT calculation software (for validation) [38]
Procedure:
- Latent Space Property Mapping:
- Encode compounds with known property values
- Train property prediction model on latent representations
- Validate prediction accuracy against experimental or DFT-calculated values [38]
Gradient-Based Optimization:
- Initialize latent vector z from prior distribution N(0,I)
- Compute gradient of target property with respect to z: ∇_z P(z)
- Iteratively update z to optimize property: z{t+1} = zt + α ∇_z P(z)
- Decode optimized latent vectors to generate candidate structures
Latent Space Interpolation:
- Select seed compounds with desired properties
- Encode compounds to obtain latent representations
- Perform linear interpolation between latent points: z' = z1 + λ(z2 - z_1)
- Decode interpolated points at regular intervals
- Validate properties of generated intermediates
Validation:
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Tools for NP-VAE Implementation
| Tool/Resource | Function | Application in NP-VAE Research |
|---|---|---|
| RDKit | Cheminformatics toolkit | Chemical structure handling, validity checking, and fingerprint generation [20] [38] |
| Tree-LSTM | Tree-structured recurrent neural network | Processing hierarchical molecular graph decompositions [20] [38] |
| ECFP (Extended Connectivity Fingerprints) | Molecular representation | Capturing circular substructures and chirality information [20] [38] |
| DrugBank Database | Pharmaceutical compound database | Source of approved drugs for training data [20] |
| Natural Product Libraries | Specialized compound collections | Source of complex natural structures for model training [20] |
| DFT Calculation Software (Q-Chem) | Quantum chemistry calculations | Validation of generated compounds' electrochemical properties [38] |
| JCESR/MP Dataset | Electrochemical property database | Training data for property prediction tasks [38] |
Discussion and Future Perspectives
NP-VAE represents a significant advancement in molecular generative modeling, specifically addressing the challenges of large, complex natural product structures with 3D complexity. Its ability to handle chirality and reconstruct large compounds with high accuracy positions it as a valuable tool for drug discovery and materials science. The integration of structural information through graph-based representations and tree-structured processing enables the model to capture essential features of complex molecules that SMILES-based approaches miss [20].
The application of NP-VAE in diverse domains, from natural product-based drug discovery to electrolyte additive design, demonstrates its versatility and robustness [20] [38]. The model's continuous latent space provides researchers with an explorable representation of chemical space, enabling rational design of novel compounds with optimized properties. The exceptional performance in predicting HOMO and LUMO values with DFT-level accuracy suggests potential for reducing computational costs in virtual screening pipelines [38].
Future developments may focus on incorporating synthetic accessibility metrics, expanding 3D conformational handling, and integrating with automated synthesis platforms. As generative models continue to evolve, NP-VAE's approach to handling structural complexity and chirality will likely inform next-generation architectures for molecular design, further bridging the gap between computational prediction and experimental realization in molecular discovery.
Conditional VAEs for Property-Guided Molecule Generation
The exploration of chemical space for novel drug candidates is a monumental challenge in pharmaceutical research, as the space of synthesizable small molecules is estimated to exceed 10³³ compounds [5]. Generative models have emerged as a principled approach to navigate this vast space efficiently. Among these, Variational Autoencoders (VAEs) have significantly influenced molecular generation due to their ability to create smooth, continuous latent spaces amenable to interpolation and optimization [5]. Conditional Variational Autoencoders (CVAEs) extend this paradigm by incorporating property vectors into both the encoder and decoder during training, enabling targeted, property-aware generation [5]. This application note details the theoretical foundation, current implementations, and experimental protocols for using CVAEs in de novo molecular design, providing researchers with practical guidance for implementing these methods in drug discovery pipelines.
Theoretical Framework: From VAE to CVAE
Variational Autoencoder (VAE) Fundamentals
The standard VAE is a directed graphical generative model that learns to reconstruct its inputs through a probabilistic encoder-decoder architecture. It assumes data X (e.g., molecular representations) is generated by an unobserved continuous random variable z (the latent representation). The encoder learns to approximate the posterior distribution q_φ(z|X), mapping inputs to a latent distribution, while the decoder learns the likelihood distribution P_θ(X|z), reconstructing data from latent points [39]. The model is trained to minimize an objective function consisting of two terms: a reconstruction loss (expected negative log-likelihood) that encourages accurate input reconstruction, and a Kullback-Leibler (KL) divergence that regularizes the learned latent distribution q_φ(z|X) towards a prior P(z), typically a standard Gaussian distribution [39].
Conditional VAE (CVAE) Extension
The fundamental limitation of standard VAEs for controlled generation is the inability to specify desired properties in the generated output. CVAEs address this by incorporating conditional information c (e.g., target molecular properties) into both the encoding and decoding processes [21] [39]. The objective function of the VAE is modified accordingly:
- Standard VAE Objective:
E[log P(X|z)] - D_KL[Q(z|X) || P(z)] - CVAE Objective:
E[log P(X|z, c)] - D_KL[Q(z|X, c) || P(z|c)][21]
This modification means the encoder learns q_φ(z|X, c),
and the decoder learns P_θ(X|z, c). During generation, sampling from the prior P(z|c) conditioned on the desired properties c and feeding it to the decoder yields molecules with those target properties [39]. This architecture provides direct control over output characteristics, a crucial capability for rational molecular design.
Current CVAE Architectures for Molecular Generation
Recent research has produced several advanced CVAE implementations tailored to molecular generation challenges, including interpretability, validity, and posterior collapse.
Table 1: Advanced CVAE Architectures for Molecular Generation
| Model Name | Key Innovation | Molecular Representation | Target Properties | Performance Highlights |
|---|---|---|---|---|
| STAR-VAE [5] | Transformer encoder & autoregressive decoder; LoRA for fine-tuning | SELFIES | Docking scores, binding affinity | Matches/exceeds baselines on GuacaMol & MOSES; shifts docking score distributions toward stronger binding |
| ICVAE [40] | Establishes linear mapping between latent variables & molecular properties | SMILES | HBA, HBD, MW, LogP, SAS, QED, TPSA | Enables precise property control via direct latent space manipulation; provides interpretable latent dimensions |
| PCF-VAE [3] | Mitigates posterior collapse; uses GenSMILES representation | GenSMILES (enhanced SMILES) | MW, LogP, TPSA | 98.01% validity (D=1); 100% uniqueness; 95.01% novelty (D=3) on MOSES |
| DiffGui [41] | Target-aware 3D generation with bond diffusion & property guidance | 3D Graph (Atom coordinates & types) | Binding affinity, QED, SA, LogP, TPSA | State-of-the-art on PDBbind; generates molecules with high affinity & rational 3D structure |
| Base CVAE [21] | Conditions both encoder & decoder on property vector | SMILES | MW, LogP, HBD, HBA, TPSA | Proof-of-concept for multi-property control; property adjustment without structural degradation |
Key Architectural Trends
- Representation Evolution: While early models used SMILES strings [21], recent approaches like STAR-VAE use SELFIES to guarantee 100% syntactic validity [5]. 3D methods like DiffGui represent molecules as graphs with atomic coordinates [41].
- Conditioning Mechanisms: Advanced models incorporate conditioning at multiple stages. STAR-VAE applies a consistent conditioning signal to the latent prior, inference network, and decoder [5].
- Interpretability Advances: ICVAE introduces a modified loss function that correlates latent values directly with molecular properties, creating a semantically structured latent space where dimensions correspond to specific properties like molecular weight (MW) or hydrogen bond acceptors (HBA) [40].
Experimental Protocols
Data Preprocessing and Molecular Representation
Protocol 1: SMILES/SELFIES Preparation
- Data Sourcing: Curate large-scale datasets from public repositories like PubChem [5] or ZINC [21]. Apply drug-likeness filters (e.g., MW ≤ 600, HBD ≤ 5, HBA ≤ 10) for pharmaceutical relevance [5].
- Standardization: Canonicalize SMILES strings for unique molecular representation [21]. For SELFIES, ensure all strings conform to valid syntax rules [5].
- Tokenization: Convert strings into machine-readable format. Pad sequences with start/end tokens (e.g., 'E') to uniform length [40] [21]. Transform each character into a one-hot vector [40] [21].
- Embedding: Transform one-hot vectors into dense embedding vectors (e.g., size 300) to capture semantic relationships between tokens [21].
Protocol 2: Property Conditioning Vector Construction
- Property Selection: Choose relevant molecular properties (e.g., MW, LogP, HBD, HBA, TPSA, QED, synthetic accessibility) [41] [21].
- Normalization: Continuously valued properties (MW, LogP, TPSA) are normalized, typically to the range [-1.0, 1.0] [21].
- Discrete Encoding: Integer-valued properties (HBD, HBA) are expressed using one-hot vectors [21].
- Vector Assembly: Concatenate all processed property values into a unified condition vector
c[21].
Model Training
Protocol 3: CVAE Training with Structural Constraints
- Architecture Selection: Choose encoder-decoder architecture. Recurrent Neural Networks (RNNs) with LSTM cells are common for sequence data [21], while Transformer-based encoders and decoders offer modern alternatives [5].
- Condition Integration: Concatenate the condition vector
cwith the embedded input matrix before the encoder and with the latent vectorzbefore each decoder step [21]. - Loss Optimization: Minimize the CVAE objective function using gradient-based optimizers like Adam [42].
- Validation: Monitor reconstruction accuracy and latent space regularization. Use early stopping to prevent overfitting [42].
Protocol 4: Interpretable CVAE (ICVAE) Training
- Modified Loss Function: Implement a loss function that penalizes deviations from a linear relationship
z = τc + ϵbetween latent valueszand property labelsc, whereτscales the latent range andϵrepresents stochasticity [40]. - Latent-Property Alignment: Explicitly associate specific latent dimensions with target molecular properties during training [40].
- Convergence Verification: Ensure latent space distributions for each property value show continuous linear relationships [40].
Conditional Generation and Evaluation
Protocol 5: Property-Guided Sampling
- Target Specification: Define desired property values for generated molecules.
- Conditioning: Construct the condition vector
cusing Protocol 2. - Sampling: For standard CVAEs, sample latent vector
zfrom the priorP(z|c). For ICVAE, directly set latent coordinates based on the linear property mapping [40]. - Decoding: Feed
zandcto the decoder to generate molecular representations. - Stochastic Write-Out: For sequence-based models, perform multiple sampling steps (e.g., 100 times) per latent vector, selecting valid, non-duplicated molecules for analysis [21].
Protocol 6: Molecular Output Validation
- Validity Check: Use toolkits like RDKit to validate chemical syntax and structure of generated molecules [3] [21].
- Property Calculation: Compute actual properties of generated molecules and compare with target values.
- Diversity Assessment: Calculate internal diversity metrics (intDiv, intDiv2) to evaluate structural variety [3].
- Novelty Evaluation: Determine novelty by comparing generated molecules with training set structures.
- Advanced Validation: For target-aware generation, compute docking scores or other binding affinity measures [5] [41].
Workflow Visualization
End-to-End CVAE Workflow for Molecular Generation
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Software Tools and Resources for Molecular CVAE Implementation
| Tool/Resource | Type | Primary Function | Application in CVAE Research |
|---|---|---|---|
| RDKit [21] | Cheminformatics Library | Molecular validation & property calculation | Check SMILES/SELFIES validity; compute properties (MW, LogP, HBD, HBA, TPSA) for generated molecules |
| PubChem [5] | Chemical Database | Source of training data | Curate large-scale (e.g., 79M molecules), drug-like datasets for pretraining |
| ZINC [21] | Virtual Compound Library | Source of training data | Provide molecular structures for model training and benchmarking |
| SELFIES [5] | Molecular Representation | Syntax-guaranteed string representation | Ensure 100% syntactic validity in generated molecular strings |
| MOSES/GuacaMol [5] | Benchmarking Platforms | Standardized evaluation | Compare model performance on validity, uniqueness, novelty, diversity |
| Low-Rank Adaptation (LoRA) [5] | Fine-tuning Technique | Parameter-efficient adaptation | Enable fast model adaptation to new properties with limited data |
| TensorFlow/PyTorch [42] | Deep Learning Frameworks | Model implementation | Build, train, and deploy CVAE architectures |
Conditional VAEs represent a powerful framework for property-guided molecular generation, bridging deep generative modeling with practical drug discovery needs. Modern implementations like STAR-VAE, ICVAE, and PCF-VAE demonstrate significant advances in scalability, interpretability, and robustness. By following the standardized protocols and utilizing the toolkit outlined in this document, researchers can effectively implement these methods to explore chemical space more efficiently and generate novel molecular structures with precisely controlled properties. The continued evolution of CVAE architectures promises further enhancements in 3D-aware generation, multi-property optimization, and integration with experimental validation pipelines.
Variational Autoencoders (VAEs) have emerged as a transformative deep learning architecture for de novo molecular design, enabling researchers to navigate the vast chemical space of drug-like compounds (estimated at 10^23 to 10^60 molecules) with unprecedented precision [3]. By mapping molecular structures into a continuous latent space, VAEs facilitate the generation of novel compounds optimized for specific therapeutic objectives, addressing a fundamental challenge in modern pharmaceutical development [43]. This Application Note details three specialized VAE architectures—PCF-VAE, ScafVAE, and SmilesGEN—that demonstrate the practical implementation of this technology across distinct drug discovery paradigms, from overcoming posterior collapse to enabling scaffold-aware generation and phenotype-informed design.
Case Study 1: PCF-VAE for Posterior Collapse-Free Molecular Generation
The PCF-VAE (Posterior Collapse Free Variational Autoencoder) framework addresses a critical limitation in conventional VAEs: the tendency to ignore latent space sampling, resulting in low-diversity molecular output [3]. This approach introduces novel reparameterization of the VAE loss function alongside simplified molecular representations to enhance robustness and diversity in generated compounds.
Experimental Protocol
Step 1: Molecular Representation Preprocessing
- Convert SMILES strings to GenSMILES representations to preserve intrinsic semantic information while reducing complexity
- Integrate key molecular properties (molecular weight, LogP, TPSA) directly into GenSMILES representations
- Implement data augmentation through multiple valid SMILES strings per molecule
Step 2: Model Architecture Specifications
- Implement modified loss function with enhanced regularization terms
- Incorporate a dedicated diversity layer between latent space and decoder
- Configure encoder-decoder architecture with GRU or transformer blocks
Step 3: Training Procedure
- Pre-train on large-scale molecular datasets (e.g., ZINC, ChEMBL)
- Employ progressive training with increasing diversity parameters
- Validate reconstruction accuracy and latent space organization
Step 4: Molecular Generation
- Sample from well-regulated latent space with controlled diversity parameters
- Decode latent vectors to generate novel molecular structures
- Validate chemical correctness and uniqueness via automated checks
Key Performance Metrics
Table 1: Quantitative Performance of PCF-VAE on MOSES Benchmark
| Metric | Diversity Level 1 | Diversity Level 2 | Diversity Level 3 |
|---|---|---|---|
| Validity | 98.01% | 97.10% | 95.01% |
| Novelty | 93.77% | 94.71% | 95.01% |
| Uniqueness | 100% | 100% | 100% |
| Internal Diversity (intDiv2) | 85.87-86.33% | 85.87-86.33% | 85.87-86.33% |
Case Study 2: ScafVAE for Multi-Objective Drug Design
ScafVAE represents a scaffold-aware variational autoencoder designed for in silico graph-based generation of multi-objective drug candidates [43]. By integrating bond scaffold-based generation with perplexity-inspired fragmentation, it expands accessible chemical space while preserving high chemical validity, enabling the design of dual-target therapeutics against complex diseases like cancer.
Experimental Protocol
Step 1: Molecular Graph Processing
- Represent molecules as graphs with atom nodes and bond edges
- Initialize node features using one-hot encoding of atom elements
- Initialize edge features using one-hot encoding of bond types
Step 2: Perplexity-Inspired Fragmentation
- Utilize pre-trained masked graph model as perplexity estimator
- Calculate bond perplexity scores to identify fragmentation points
- Generate molecular fragments based on uncertainty metrics
Step 3: Bond Scaffold-Based Generation
- Assemble bond scaffolds without specifying atom types
- Decorate scaffolds with appropriate atom types iteratively
- Validate molecular validity at each assembly step
Step 4: Multi-Objective Optimization
- Train surrogate models on latent space for property prediction
- Simultaneously optimize for multiple properties (binding affinity, toxicity, drug-likeness)
- Sample latent vectors from optimized regions for candidate generation
Step 5: Experimental Validation
- Conduct molecular docking against target proteins
- Measure binding affinity experimentally or computationally
- Perform molecular dynamics simulations for stability assessment
Key Performance Metrics
Table 2: ScafVAE Performance on Multi-Objective Design Tasks
| Optimization Objective | Performance Metric | Result |
|---|---|---|
| Dual-Target Binding | Docking Score Improvement | 25-40% vs. baseline |
| Drug-Likeness | QED Score | >0.7 |
| Synthetic Accessibility | SA Score | <3.5 |
| ADMET Properties | Prediction Accuracy | 85-92% |
| Chemical Validity | Valid Structures | >95% |
Case Study 3: SmilesGEN for Phenotypic Drug Discovery
SmilesGEN employs a dual-channel VAE architecture to generate drug-like molecules capable of inducing desirable phenotypic changes [44]. By jointly modeling the interplay between drug perturbations and transcriptional responses in a common latent space, it bridges the gap between phenotypic screening and target-based design approaches.
Experimental Protocol
Step 1: Data Collection and Preprocessing
- Compile drug-induced transcriptional response data (e.g., L1000 platform)
- Curate paired pre-treatment and post-treatment expression profiles
- Assemble molecular structures with corresponding phenotypic profiles
Step 2: Dual-Channel VAE Architecture
- Implement molecule VAE (SmilesNet) with GRU backbone for SMILES encoding
- Implement expression profile VAE (ProfileNet) with feed-forward layers
- Establish shared latent space with cross-channel regularization
Step 3: Model Training
- Pre-train SmilesNet on large-scale SMILES descriptors
- Train ProfileNet to reconstruct expression profiles
- Optimize joint latent space to encode drug perturbative effects
- Enforce constraint that encoded post-treatment profiles adjusted by molecular representation should restore pre-treatment state
Step 4: Phenotype-Informed Molecular Generation
- Encode desired phenotypic profiles into latent space
- Decode through SmilesNet to generate candidate molecules
- Validate generated structures for chemical validity and novelty
Step 5: Experimental Validation
- Conduct high-throughput phenotypic screening (e.g., Cell Painting)
- Measure transcriptional responses to generated compounds
- Compare induced profiles to desired phenotypic signatures
Key Performance Metrics
Table 3: SmilesGEN Performance on Phenotype-Informed Generation
| Evaluation Metric | SmilesGEN | Baseline Models |
|---|---|---|
| Validity | 92.4% | 84.7% |
| Uniqueness | 98.2% | 95.1% |
| Novelty | 90.7% | 82.3% |
| Tanimoto Similarity to Known Ligands | 0.72 | 0.58 |
| Phenotypic Signature Match | 85% | 70% |
Research Reagent Solutions
Table 4: Essential Research Reagents and Computational Tools
| Reagent/Software | Specifications | Application Function |
|---|---|---|
| GenSMILES Converter | Custom Python package | Simplifies SMILES complexity for robust model input |
| MOSES Benchmark | Platform: Python | Standardized evaluation of molecular generation models |
| L1000 Expression Profiling | Platform: Luminex | High-throughput gene expression measurement for phenotypic signatures |
| Cell Painting Assay | Kit: Multiple fluorophores | High-content morphological profiling for phenotypic screening |
| RDKit | Version: 2020.09+ | Cheminformatics toolkit for molecular manipulation and validation |
| AutoDock Vina | Version: 1.2.0 | Molecular docking for binding affinity prediction |
| GROMACS | Version: 2020+ | Molecular dynamics simulations for binding stability analysis |
| OpenPhenom | Model: CA-MAE ViT-S/16 | Feature extraction from Cell Painting images |
Workflow Visualization
VAE Architectures for Targeted Drug Discovery - This workflow illustrates the three specialized VAE approaches for different therapeutic objectives, converging through experimental validation to optimized drug candidates.
ScafVAE Bond Scaffold Generation Protocol - This detailed workflow shows the complete bond scaffold-based generation process from molecular graph input to valid multi-objective drug candidate output.
The case studies presented demonstrate how specialized VAE architectures are addressing critical challenges in targeted drug discovery. PCF-VAE overcomes fundamental limitations in molecular diversity, ScafVAE enables rational design of multi-target therapeutics, and SmilesGEN bridges phenotypic screening with molecular generation. As these technologies mature, their integration into automated drug discovery platforms promises to significantly compress development timelines from years to months while increasing success rates in clinical translation [45] [46]. The experimental protocols and analytical frameworks provided herein offer researchers comprehensive guidelines for implementing these advanced molecular generation approaches in their own drug discovery pipelines.
Overcoming Key Challenges: Posterior Collapse, Novelty, and Validity
Identifying and Mitigating Posterior Collapse in VAEs
In the context of molecular generation research, the Variational Autoencoder (VAE) has emerged as a prominent framework for de novo drug design. A VAE learns to compress high-dimensional molecular representations (e.g., SMILES strings or molecular graphs) into a low-dimensional latent space, and then reconstruct them via a decoder [47] [20]. The model is trained by maximizing the Evidence Lower Bound (ELBO), which balances a reconstruction loss and a Kullback-Leibler (KL) divergence term that regularizes the latent space [47].
A major limitation in this framework is the posterior collapse phenomenon (also known as KL vanishing) [48]. This occurs when the model's powerful decoder ignores the latent variables z sampled from the approximate posterior qϕ(z|x). The posterior distribution then becomes indistinguishable from the prior p(z), causing the latent variables to carry no information about the input data [49] [48]. For molecular generation, this is catastrophic: the model fails to learn meaningful molecular representations, and generated molecules lack diversity and critical property optimizations [3]. The decoder relies solely on its autoregressive capabilities (e.g., predicting the next character in a SMILES string based on previous ones), effectively reducing the VAE to a simpler autoregressive model [48].
Diagnosing Posterior Collapse
Quantitative Diagnostics
Researchers should monitor the following metrics during VAE training to identify posterior collapse.
Table 1: Key Quantitative Metrics for Diagnosing Posterior Collapse
| Metric | Healthy VAE | Collapsed VAE | Calculation/Interpretation |
|---|---|---|---|
| KL Divergence | Stable, positive value | Approaches zero | ( D{KL}[q\phi(z|x) | p(z)] ); Near zero indicates collapse [47] [48]. |
| Active Units (AUs) | High number | Low number | Dimensions in z where ( \text{Cov}(μ(x)) > \delta ); measures latent space utilization [48]. |
| Reconstruction Loss | Decreases and stabilizes | Decreases rapidly | ( \mathbb{E}{q\phi(z|x)}[\log p_\theta(x|z)] ); Very low loss with high KL can signal a powerful decoder ignoring z [49]. |
| Mutual Information | High | Low | ( I(x; z) ); Measures dependence between data and latent variables [48]. |
A Phase Transition Perspective
Recent theoretical work frames posterior collapse as a phase transition governed by data structure and model hyperparameters [47]. A key finding is that for a deep Gaussian VAE, collapse initiates when the decoder's variance exceeds the largest eigenvalue of the data covariance matrix [47]. At this critical point, the KL divergence exhibits non-analytic behavior, confirming its phase transition nature [47]. This provides a theoretical criterion for diagnosing collapse risk before training begins.
Mitigation Strategies for Molecular VAEs
Several strategies have been developed to mitigate posterior collapse, which can be categorized and applied to molecular generation tasks.
Table 2: Strategies to Mitigate Posterior Collapse
| Strategy | Mechanism | Example Implementations in Molecular VAEs | Pros/Cons |
|---|---|---|---|
| Architectural & Objective Function Changes | Modifies the VAE objective to prevent the KL term from vanishing. | β-VAE [50], Conditional VAE (CVAE) [21], PCF-VAE [3] | Pros: Often effective; CVAE enables property control [21]. Cons: Can require extensive tuning (e.g., of β). |
| Training Schedule Techniques | Adjusts the training dynamics to encourage latent variable use early in training. | KL Annealing [48], KL Weight Dropout [48] | Pros: Simple to implement. Cons: May not suffice for highly expressive decoders; annealing schedule is sensitive [48]. |
| Input Manipulation | Reduces the decoder's autoregressive power, forcing it to rely on the latent variable. | Word/Token Dropout [48], DVAE Model [48] | Pros: Very effective for text/SMILES-based models. Cons: Risks under-utilizing the decoder if over-applied [48]. |
Protocol: Mitigating Collapse with the DVAE Framework for SMILES Generation
The Dropout VAE (DVAE) introduces a dual-path decoder during training to combat collapse in text modeling, which is directly applicable to SMILES string generation [48].
Workflow Overview:
Procedure:
- Input Processing: For each SMILES string
xin a training batch, create two copies:- Path A: The original SMILES string
x. - Path B: A corrupted version
x_corruptedwhere a random subset of tokens (e.g., 20-40%) is replaced with a generic<unk>token [48].
- Path A: The original SMILES string
- Encoding: The encoder
q_ϕ(z|x)processes the originalxto produce the parameters of the posterior distribution (meanμand varianceσ²). A latent variablezis sampled via the reparameterization trick [48]. - Dual-Path Decoding: The same decoder
p_θ(x|z)and latent variablezare used for both paths.- The decoder processes
Path Ato compute reconstruction lossL_rec_A. - The decoder processes
Path Bto compute reconstruction lossL_rec_B.
- The decoder processes
- Loss Calculation: The total reconstruction loss is the average of
L_rec_AandL_rec_B. This is combined with the KL divergence to form the ELBO objective [48]: ( \mathcal{L}{\text{DVAE}} = \frac{1}{2}[\log p\theta(x|z) + \log p\theta(x{\text{corrupted}}|z)] - D{KL}[q\phi(z|x) \| p(z)] ) - Stopping Strategy & Fine-Tuning: Once the model converges (e.g., validation reconstruction loss plateaus), disable
Path Band continue training for a few epochs with onlyPath A. This ensures the decoder fully utilizes its expressive power once the latent variables are actively used [48].
Protocol: Using β-VAE and Property Control for Molecular Generation
The β-VAE and Conditional VAE (CVAE) frameworks are highly effective for molecular generation, where controlling properties is essential [50] [21].
Workflow Overview:
Procedure for Conditional β-VAE:
- Model Architecture:
- The input is a SMILES string and a condition vector
ccontaining target molecular properties (e.g., Molecular Weight (MW), LogP, HBD, HBA, TPSA) [21]. - The encoder network takes the concatenated input
[x, c]and outputs parameters forq_ϕ(z|x, c). - The decoder network takes the concatenated input
[z, c]and outputs the probability distributionp_θ(x|z, c)[21].
- The input is a SMILES string and a condition vector
- Objective Function: The ELBO is modified to include the condition
cand the β coefficient: ( \mathcal{L}{\text{Cβ-VAE}} = \mathbb{E}{q\phi(z|x,c)}[\log p\theta(x|z,c)] - \beta \cdot D{KL}[q\phi(z|x,c) \| p(z|c)] ) whereβ > 1punishes the KL term more heavily, encouraging a more disentangled and robust latent space that is less prone to collapse [50]. - Training: The model is trained to reconstruct the input SMILES
xwhile the latent space is structured by the KL term and explicitly conditioned on the propertiesc. This direct conditioning prevents the model from ignoring the latent variables, as they are necessary for generating molecules with the specified properties [21]. - Generation: To generate novel molecules with desired properties, sample a latent vector
zfrom the priorp(z)and concatenate it with the target condition vectorcbefore passing it to the decoder [21].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Item Name | Function/Description | Application in Molecular VAE Research |
|---|---|---|
| ZINC Dataset | A publicly available curated library of commercially available chemical compounds. | Primary source of training data for small-molecule generative models [21]. |
| MOSES Benchmark | A benchmarking platform for molecular generation models. | Standardized evaluation of model performance (e.g., validity, uniqueness, novelty, diversity) [3]. |
| RDKit | Open-source cheminformatics software. | Calculates molecular properties (e.g., LogP, TPSA), checks SMILES validity, and handles molecular representations [20] [21]. |
| GenSMILES | A preprocessed and simplified version of SMILES notation. | Reduces complexity of SMILES strings, improving model learning and generation of valid structures [3]. |
| PCF-VAE Framework | A VAE variant specifically designed to be "Posterior Collapse Free". | Generates a high diversity and validity rate of molecules, as validated on the MOSES benchmark [3]. |
| NP-VAE Framework | A graph-based VAE for handling large, complex molecules (e.g., natural products). | Constructs chemical latent spaces from large molecular structures and incorporates chirality (3D complexity) [20]. |
Strategies for Improving Generation Validity and Novelty
In the field of de novo molecular design, variational autoencoders (VAEs) have emerged as a powerful framework for exploring the vast chemical space. A VAE is a probabilistic generative model that learns to map molecules into a continuous, low-dimensional latent space and decode them back into molecular structures [51] [52]. This capability enables the generation of novel molecular entities with tailored properties. However, two persistent challenges have limited their practical utility: generation validity (producing syntactically and chemically valid structures) and novelty (generating molecules that are both unique and diverse compared to the training data) [3] [53]. This application note details proven strategies to overcome these challenges, providing researchers with practical methodologies to enhance VAE performance for drug discovery applications.
Foundational Concepts: VAEs in Molecular Generation
A VAE consists of three core components: an encoder that maps an input molecule to a probability distribution in latent space (parameterized by a mean μ and variance σ²), a latent space from which points are sampled, and a decoder that reconstructs the molecule from the sampled latent point [52]. The model is trained by optimizing a loss function comprising a reconstruction term (ensuring input fidelity) and a KL divergence term (regularizing the latent space to resemble a prior distribution, typically a standard Gaussian) [51] [52].
The standard autoencoder framework faces a critical limitation for generation: its deterministic encoding creates a disjointed latent space with significant gaps. When the decoder encounters points from these unexplored regions, it often produces invalid outputs [54]. The VAE's probabilistic approach and KL divergence loss work together to create a more continuous and structured latent space, enabling meaningful sampling and generation of novel, valid structures [54] [51].
Strategy 1: Advanced Molecular Representations
The choice of molecular representation fundamentally impacts a VAE's ability to learn valid chemical structures.
SELFIES Representation
The SELFIES (Self-Referencing Embedded Strings) representation guarantees 100% syntactic validity for all token sequences by design. This is achieved through a grammar that ensures every generated string corresponds to a chemically valid molecule, making it particularly well-suited for sequence-based generative modeling at scale [5]. In the STAR-VAE model, the use of SELFIES was instrumental in achieving high validity rates during generation [5].
GenSMILES Representation
The GenSMILES representation simplifies the complexity of standard SMILES strings while preserving semantic molecular information. This transformation helps the model learn long-range dependencies within the string and reduces the incidence of invalid outputs. Furthermore, GenSMILES can be augmented with molecular descriptors such as molecular weight, LogP, and TPSA, conditioning the VAE to generate molecules that meet specific property criteria [3].
Graph-Based Representations
Graph-based representations explicitly encode atoms as nodes and bonds as edges, directly capturing molecular topology. The Transformer Graph VAE (TGVAE) utilizes this representation to more effectively model complex structural relationships than string-based methods, leading to improved generation of diverse and valid molecules [4].
Table 1: Comparison of Molecular Representations for VAEs
| Representation | Core Principle | Impact on Validity | Impact on Novelty |
|---|---|---|---|
| SELFIES | Grammar-based rules ensure syntactic correctness. | Guarantees 100% syntactic validity [5]. | Enables exploration of novel structures by removing validity constraints. |
| GenSMILES | Simplified SMILES with integrated properties. | Reduces complexity, improving learning and validity [3]. | Conditions generation for novel molecules with desired properties. |
| Graph-Based | Direct encoding of atomic connectivity. | Avoids syntactic invalidity; ensures structurally sound molecules [4]. | Captures complex structural patterns, supporting diverse generation. |
Strategy 2: Architectural and Formulation Innovations
Modernizing the VAE architecture and its probabilistic formulation is key to enhancing performance.
Transformer-Based Architectures
Replacing traditional recurrent neural networks (RNNs) with Transformer-based encoder-decoders captures long-range dependencies in molecular sequences more effectively. The STAR-VAE framework, which employs a bi-directional Transformer encoder and an autoregressive Transformer decoder, demonstrates that this scalable architecture, when trained on large datasets (e.g., 79 million drug-like molecules from PubChem), achieves competitive performance on standard benchmarks like GuacaMol and MOSES [5].
Mitigating Posterior Collapse
Posterior collapse occurs when the model fails to use the latent space meaningfully, causing the generated molecules to lack diversity. The PCF-VAE (Posterior Collapse Free VAE) addresses this by reparameterizing the VAE loss function. This approach successfully mitigates posterior collapse, resulting in a more informative latent space and the generation of a greater variety of valid molecules, as evidenced by its high validity and uniqueness scores on the MOSES benchmark [3].
Conditional Generation Formulations
A principled conditional latent-variable formulation allows for property-guided generation. In this setup, a property predictor provides a conditioning signal that is consistently applied to the latent prior, the inference network, and the decoder. This enables controlled generation towards molecules with specific, desired attributes, effectively shifting the distribution of generated molecules toward improved property profiles, such as stronger predicted binding affinities for protein targets [5].
Strategy 3: Advanced Training and Optimization Techniques
Refining the training process and optimizing within the latent space further boost validity and novelty.
Multi-Objective Latent Space Optimization (LSO)
Multi-objective LSO reshapes the latent space to bias the generative model towards molecules that simultaneously optimize multiple properties. One effective method involves an iterative weighted retraining scheme, where molecules in the training data are weighted based on their Pareto efficiency. This guides the model to explore regions of the latent space that correspond to Pareto-optimal molecules, pushing the Pareto front for multiple properties without relying on ad-hoc scalarization [53].
Parameter-Efficient Finetuning
Low-Rank Adaptation (LoRA) applied to both the encoder and decoder enables fast adaptation of large, pre-trained VAEs with limited property-specific data. This parameter-efficient finetuning approach allows researchers to quickly specialize a general-purpose molecular generator for specific tasks, maintaining model performance while reducing computational costs [5].
Incorporation of Correlated Descriptors
Jointly training a VAE with an auxiliary predictor for molecular descriptors that are correlated with target properties can improve the organization of the latent space. This strategy forces the model to embed relevant chemical information into the latent representations, which in turn enhances the performance of downstream property prediction tasks and can guide the generation of molecules with more favorable properties [55].
Table 2: Summary of Key Experimental Results from Literature
| Model / Strategy | Benchmark / Task | Validity (%) | Uniqueness / Novelty (%) | Internal Diversity (IntDiv2, %) |
|---|---|---|---|---|
| PCF-VAE [3] | MOSES (D=1) | 98.01 | 93.77 | 85.87 - 86.33 |
| PCF-VAE [3] | MOSES (D=2) | 97.10 | 94.71 | 85.87 - 86.33 |
| PCF-VAE [3] | MOSES (D=3) | 95.01 | 95.01 | 85.87 - 86.33 |
| STAR-VAE [5] | GuacaMol / MOSES | Matches or exceeds baselines | Matches or exceeds baselines | Latent-space analyses reveal smooth, structured representations. |
| Conditional STAR-VAE [5] | Tartarus (Docking Scores) | Shifts distribution toward stronger binders, demonstrating targeted generation. | Produces many high-scoring, diverse molecules. | Captures target-specific molecular features. |
Experimental Protocols
Protocol: Implementing a SELFIES-Based Transformer VAE
This protocol outlines the steps for implementing the STAR-VAE model [5].
Data Curation and Preprocessing
- Source: Curate a large dataset of drug-like molecules (e.g., from PubChem).
- Filtering: Apply standard drug-likeness filters (e.g., Molecular Weight ≤ 600 Da, HBD ≤ 5, HBA ≤ 10).
- Representation Conversion: Convert all molecular structures to SELFIES strings using the SELFIES library.
- Tokenization: Tokenize the SELFIES strings into a vocabulary suitable for sequence modeling.
Model Architecture Setup
- Encoder: Implement a bi-directional Transformer encoder. The output of the encoder should be mapped to two separate linear layers to produce the latent mean (μ) and log-variance (log σ²) vectors.
- Latent Sampling: Implement the reparameterization trick:
z = μ + ε * exp(0.5 * log σ²), whereεis sampled from a standard normal distribution. - Decoder: Implement an autoregressive Transformer decoder. The latent vector
zis used as the initial input to start the decoding process for generating the SELFIES string token-by-token.
Training Configuration
- Loss Function: Use the standard VAE loss:
L = L_reconstruction + β * L_KL, whereL_KLis the KL divergence between the learned latent distribution and a standard normal prior. The weightβcan be tuned. - Optimization: Use the Adam optimizer with a suitable learning rate (e.g., 1e-4) and batch size.
- Loss Function: Use the standard VAE loss:
Conditional Generation Finetuning (Optional)
- Property Predictor: Add a feed-forward property prediction network that takes the latent vector
zas input. - Conditioning Signal: Incorporate the property prediction as a conditioning signal into the prior, encoder, and decoder during training.
- Parameter-Efficient Finetuning: Use LoRA to finetune the pre-trained model on a small dataset labeled with the target property.
- Property Predictor: Add a feed-forward property prediction network that takes the latent vector
Diagram 1: SELFIES VAE workflow.
Protocol: Mitigating Posterior Collapse with PCF-VAE
This protocol is based on the PCF-VAE approach to prevent posterior collapse and enhance diversity [3].
Data Representation with GenSMILES
- Transform standard SMILES strings into GenSMILES representations to reduce complexity.
- (Optional) Compute and append key molecular descriptors (e.g., MW, LogP, TPSA) to the GenSMILES string.
Model Modification
- Implement a standard VAE architecture (e.g., with RNN or Transformer encoder/decoder).
- Reparameterize the Loss Function: Modify the VAE objective function as proposed in PCF-VAE to aggressively penalize the KL term when it falls below a threshold, forcing the model to utilize the latent space.
Diversity Enhancement
- Introduce a diversity layer between the latent space and the decoder. This layer can be designed to explicitly encourage the dispersion of latent points.
- Train the model with the modified loss function, monitoring both reconstruction accuracy and the KL divergence term to ensure it does not collapse to zero.
Diagram 2: PCF-VAE architecture.
Protocol: Multi-Objective Latent Space Optimization
This protocol describes the weighted retraining method for multi-property optimization [53].
Initial Model Pretraining
- Pretrain a VAE (e.g., JT-VAE, STAR-VAE) on a large, unlabeled dataset of molecules (e.g., ZINC).
Iterative Weighted Retraining Loop
- Step 1 - Generate & Evaluate: Use the current VAE to generate a large set of candidate molecules. Calculate their properties using relevant predictors or simulations.
- Step 2 - Pareto Ranking: Rank all molecules (both generated and the original training set) based on Pareto efficiency with respect to the multiple target properties.
- Step 3 - Assign Weights: Assign a weight to each molecule in the training set based on its Pareto rank. Higher-ranked molecules receive larger weights.
- Step 4 - Retrain Model: Retrain the VAE on the weighted training dataset. This biases the model towards the regions of latent space that produce high-performing molecules.
- Step 5 - Iterate: Repeat steps 1-4 for a fixed number of cycles or until performance converges.
Diagram 3: Multi-objective LSO workflow.
Table 3: Essential Resources for Molecular VAE Research
| Resource / Tool | Type | Primary Function in Research | Example Use Case |
|---|---|---|---|
| PubChem [5] | Data Repository | Source of large-scale, drug-like molecular structures for pre-training. | Curating a dataset of ~79 million molecules for foundational model training. |
| ZINC [55] | Database | A curated database of commercially available compounds for benchmarking and training. | Sourcing the ZINC-250k dataset for model pre-training and comparison. |
| RDKit | Cheminformatics Library | Computes molecular descriptors, handles SMILES/SELFIES conversion, and validates chemical structures. | Filtering datasets, calculating properties like LogP, and validating generated molecules. |
| SELFIES [5] | Representation Library | Python library to convert molecules to and from SELFIES strings. | Guaranteeing 100% syntactic validity of generated molecular strings. |
| MOSES [5] [3] | Benchmarking Platform | Standardized benchmark to evaluate the performance of generative models. | Comparing the validity, uniqueness, and diversity of a new model against published baselines. |
| GuacaMol [5] | Benchmarking Platform | Benchmark for goal-directed generative models, assessing property optimization capabilities. | Evaluating a model's ability to generate molecules with specific target properties. |
| LoRA [5] | Finetuning Method | Parameter-efficient finetuning technique for large models. | Adapting a pre-trained VAE to a new protein target with limited data. |
Addressing Over-Smoothing in Graph Neural Networks
In the field of molecular generation, Graph Neural Networks (GNNs) have become a cornerstone technology, particularly when integrated with Variational Autoencoders (VAEs) for de novo drug design. These models excel at capturing the complex structural relationships within molecules more effectively than traditional string-based representations [4]. However, a significant challenge known as over-smoothing can impede their performance. This phenomenon describes a condition where, as a GNN gains depth through the addition of more layers, node representations (embeddings) become increasingly similar and ultimately indistinguishable [56] [57]. In the context of molecular graphs, this leads to a loss of critical structural information, resulting in the generation of invalid, non-diverse, or undesirable molecules [3].
The message passing framework, the core operational mechanism of GNNs, is intrinsically linked to over-smoothing. In this process, each node updates its embedding by aggregating features from its neighboring nodes (the Aggregate function) and then combining this gathered information with its own features (the Update function) [56] [57]. While this mechanism allows nodes to harness information from their local graph structure, it also inherently performs a smoothing operation. As more layers are stacked, a node's receptive field expands to include more distant neighbors (e.g., second-hop neighbors). This quest for greater contextual awareness can backfire, causing node embeddings across the entire graph to converge, which severely hampers the model's ability to perform precise downstream tasks like node classification or the generation of distinct molecular structures [56] [57] [58].
It is crucial to recognize that over-smoothing is not a simple bug but rather an inherent characteristic of the message-passing paradigm [56]. For research aimed at generating novel molecular entities, mitigating this issue is not optional but essential for success. This document serves as a comprehensive set of application notes and protocols, designed to equip researchers and drug development professionals with the knowledge and tools to effectively detect, quantify, and counteract over-smoothing in GNNs, with a specific focus on applications within molecular graph generation.
Quantifying and Detecting Over-Smoothing
To systematically address over-smoothing, researchers must first be able to quantify it. Several well-established metrics allow for the tracking and diagnosis of this phenomenon during model training and evaluation.
Table 1: Metrics for Quantifying Over-Smoothing in GNNs
| Metric Name | Full Name | Primary Function | Interpretation |
|---|---|---|---|
| MAD [56] | Mean Average Distance | Measures the overall similarity of node representations in the graph. | A higher MAD value indicates greater similarity between node embeddings, signifying a higher degree of smoothness. The value typically increases with the number of GNN layers. |
| MADGap [56] | Mean Average Distance Gap | Assesses the similarity of representations across different node classes. | Quantifies the information-to-noise ratio for a node during message passing. A lower ratio suggests that a node is gathering more noise from other classes, which is detrimental to classification. |
| Information-to-Noise Ratio [58] | Information-to-Noise Ratio | Evaluates the quality of information a node receives from its neighbors during aggregation. | A declining ratio indicates that nodes from different classes are being aggregated, introducing noise and reducing the discriminative power of the embeddings. |
The application of these metrics is straightforward. By plotting metrics like MAD against the number of GNN layers, researchers can visually identify the point at which the model begins to suffer from over-smoothing, characterized by a sharp increase in MAD or a sharp decrease in MADGap [56]. This empirical analysis helps in determining the optimal depth for a GNN model before performance degradation occurs.
Mitigation Strategies and Protocols
A range of strategies has been developed to mitigate over-smoothing, enabling the construction of deeper and more powerful GNNs. The following sections detail several key approaches, complete with implementation protocols.
Architectural Innovations
Adaptive Early Embedding (AEE) with Biased DropEdge (BDE)
This combined procedure is a state-of-the-art plug-and-play method that can be integrated with standard message-passing GNNs like GCN and GAT [58].
- Core Principle of AEE: The model utilizes auxiliary networks at intermediate GNN layers to assess the classification confidence of individual node embeddings. Nodes that can be classified with high confidence at an earlier layer are allowed to "exit" or "halt" the message-passing process prematurely. This prevents their embeddings from being further smoothed by subsequent, potentially noisy, aggregations [58].
- Core Principle of BDE: This technique selectively removes edges between nodes of different classes (inter-class edges) during training. By reducing connections that are likely to introduce noisy information, BDE actively improves the information-to-noise ratio in the message-passing process [58].
Table 2: Research Reagent Solutions for AEE+BDE Implementation
| Component Name | Type/Function | Implementation Notes |
|---|---|---|
| Main GNN Backbone | Base Network | Serves as the primary feature extractor (e.g., GCN, GAT). Choose based on graph characteristics (homophily/heterophily). |
| Auxiliary Networks | Early Exit Classifiers | Small classification networks attached to intermediate layers. They determine if a node's embedding is sufficient for a confident prediction. |
| Confidence Threshold | Hyperparameter | A pre-defined confidence score from the auxiliary network that triggers an early embedding exit. Tunable based on validation set performance. |
| Edge Bias Mask | Pre-processing Filter | A mask applied to the adjacency matrix to selectively drop inter-class edges based on available label information. |
Experimental Protocol for AEE+BDE:
- Model Setup: Begin with a deep GNN backbone (e.g., a 10-layer GCN). After each of the first L-1 layers, attach an auxiliary classifier network.
- Training Loop: a. Forward Pass: For a given node, propagate its features through the main GNN layers. b. Early Embedding Check: After each intermediate layer, pass the node's current embedding through the corresponding auxiliary classifier. c. Exit Decision: If the classifier's confidence score for any class exceeds a pre-set threshold (e.g., 0.9), the node's final embedding is taken from this layer. It is excluded from further aggregation in deeper layers. d. Loss Calculation: The total loss is a weighted sum of the losses from all auxiliary classifiers and the final main classifier.
- BDE Integration: During each training epoch, apply the Biased DropEdge procedure by sampling a modified adjacency matrix where a portion of inter-class edges have been randomly removed.
- Inference: Use the subset of auxiliary networks (and the final layer) that demonstrated the best accuracy on the validation set to perform the early exit decisions.
Weight Reparameterization (WeightRep)
Challenging the traditional narrative that over-smoothing is an inevitable consequence of graph propagation, recent research suggests it may be more of a learning disability [59]. This approach posits that with properly learned weights, vanilla GNNs can, in theory, avoid over-smoothing entirely.
- Core Principle: The WeightRep method reparameterizes the weights of the GNN to adaptively maintain them close to an "ideal" state throughout the learning process. This prevents the weights from causing the node features to collapse into an indistinguishable state, even after many propagation steps [59].
Experimental Protocol for WeightRep:
- Model Selection: Choose a standard GNN architecture (e.g., vanilla GCN).
- Reparameterization: Replace the standard weight matrices of the GNN with a reparameterized version. The exact form of this reparameterization is model-specific but is designed to constrain the learning dynamics to avoid pathological regions that lead to over-smoothing.
- Training: Train the model using standard backpropagation and optimization techniques. The reparameterization acts as an implicit regularizer, guiding the weight learning.
- Validation: Monitor task-specific performance (e.g., node classification accuracy) and over-smoothing metrics (e.g., MAD) across layers to confirm the mitigation effect.
Regularization and Feature Transformation Techniques
Non-Linear Feature Transformation
A simple yet effective technique to increase the expressive power of each GNN layer and combat over-smoothing is the insertion of non-linear feedforward neural network layers (e.g., MLPs) within each GNN layer [56].
- Core Principle: This technique applies a non-linear transformation to the node embeddings at each layer before they are passed to the next message-passing step. This enhances the model's capacity to learn complex, non-linear relationships in the data, thereby slowing down the convergence of node representations [56].
- Protocol: Within the
Updatefunction of a GNN layer, after aggregating neighbor information, pass the combined embedding through a small MLP with a non-linear activation function (e.g., ReLU) before outputting the final embedding for that layer.
The Over-Smoothing and Over-Squashing Trade-off
When designing deep GNNs, it is critical to be aware of another related issue: over-squashing. This phenomenon occurs when a node's receptive field becomes too large, and information from an exponentially growing number of neighbor nodes must be compressed into a fixed-size node embedding. This can lead to a bottleneck, where distant structural information is lost [60] [61].
Notably, a trade-off exists between over-smoothing and over-squashing [60]. Techniques that enhance the sharpness of node features (e.g., high-pass filters) to fight over-smoothing can make the model more susceptible to over-squashing, and vice-versa. Therefore, a holistic approach is necessary. Models like the Multi-Scaled Heat Kernel based GNN (MHKG) [60] have been proposed to unify the analysis of both problems and offer a more balanced solution under mild conditions. For molecular generation tasks, evaluating both over-smoothing metrics and the model's ability to capture long-range dependencies is recommended.
Connecting GNN and VAE Oversmoothing in Molecular Generation
In molecular generation, the GNN is often used as the encoder within a VAE framework. In this context, the over-smoothing of node embeddings in the GNN encoder can directly contribute to a related problem in the VAE known as posterior collapse [4] [3] [62].
- The Link: If the GNN encoder produces over-smoothed, uninformative latent representations (posteriors) for all input molecules, the VAE's decoder learns to ignore the latent space and generates outputs based solely on its own biases. This results in a lack of diversity and controllability in the generated molecules [3].
- Mitigation Strategy (PCF-VAE): The Posterior Collapse Free VAE (PCF-VAE) approach addresses this by reparameterizing the VAE's loss function and simplifying the molecular representation (e.g., transforming SMILES into GenSMILES) to reduce complexity. A key innovation is the inclusion of a diversity layer between the latent space and the decoder, which explicitly controls the diversity and validity of the generated molecules [3].
Experimental Protocol for TGVAE in Molecular Design:
The Transformer Graph VAE (TGVAE) model combines a transformer, GNN, and VAE for generative molecular design [4].
- Data Preparation: Represent molecules as graphs (atoms as nodes, bonds as edges).
- Model Architecture:
- Encoder: A GNN processes the molecular graph to generate initial node embeddings, which are then aggregated into a graph-level representation.
- Latent Space: The graph-level representation is projected into the mean and variance vectors of the VAE's latent distribution.
- Decoder: A transformer decoder generates a sequential representation (e.g., a SMILES string) of the molecule from a sample taken from the latent distribution.
- Training: Train the model to maximize the reconstruction likelihood of the input molecules while regularizing the latent space with the Kullback–Leibler (KL) divergence term. Specific regularizers must be applied to prevent posterior collapse [4].
- Generation: Sample new points from the latent space and use the transformer decoder to generate novel molecular structures.
Addressing over-smoothing is a critical step toward building robust and deep GNNs for advanced molecular generation. By leveraging the quantitative metrics and mitigation protocols outlined in this document—such as Adaptive Early Embedding, Weight Reparameterization, and specialized VAE architectures—researchers and drug development professionals can significantly enhance the performance and reliability of their models. A successful strategy often involves a combination of these techniques, coupled with a careful analysis of the inherent trade-offs, to generate diverse, novel, and valid molecular structures that push the boundaries of drug discovery.
The field of de novo molecular design is undergoing a revolutionary transformation, driven by advances in deep learning and sophisticated molecular representations. Within this landscape, variational autoencoders (VAEs) have emerged as a powerful framework for navigating the vast chemical space, which is estimated to contain up to 10⁶⁰ drug-like molecules [20]. The efficacy of these models is profoundly dependent on the molecular representations they utilize. Traditional Simplified Molecular-Input Line-Entry System (SMILES) representations, while prevalent, suffer from significant validity issues, often generating syntactically or semantically invalid structures [63]. This application note examines the evolution beyond SMILES to advanced representations including GenSMILES, SELFIES, graph-based, and three-dimensional (3D) structures, framing them within the context of a broader thesis on VAE for molecule generation research. We provide a comprehensive technical overview, including quantitative performance comparisons, detailed experimental protocols, and essential toolkits for researchers and drug development professionals aiming to implement these cutting-edge approaches.
Molecular Representation Paradigms: A Quantitative Comparison
The choice of molecular representation fundamentally shapes the performance, validity, and applicability of VAE-based generative models. The following table summarizes the key characteristics, advantages, and limitations of contemporary representation schemes.
Table 1: Comparison of Advanced Molecular Representations for VAEs
| Representation | Core Principle | Reported Validity (%) | Key Advantages | Inherent Limitations |
|---|---|---|---|---|
| GenSMILES [63] [3] | Replaces paired parentheses/branches with single notations and digits to denote length. | >90 (across multiple datasets) | Addresses both syntactic and semantic issues; improves validity and diversity without long dependencies. | Requires conversion to/from standard SMILES; less human-readable. |
| SELFIES [5] | Uses self-referencing rules to guarantee 100% syntactic validity for any string. | ~100 (syntactic) | Guarantees syntactic validity; robust for large-scale training (e.g., on 79M molecules). | Lower information density; all symbols are bracketed, reducing readability. |
| Graph-Based [20] [4] | Represents molecules as graphs (atoms as nodes, bonds as edges). | ~100 (structural) | Naturally captures molecular topology; inherently ensures structural validity. | Computational complexity increases with molecular size. |
| 3D Structures [64] | Encodes atom types, bonds, and 3D Cartesian coordinates. | 100 (atom/bond accuracy) | Captures stereochemistry and spatial relationships; essential for structure-based design. | Requires handling of rotational/translational equivariance; higher data complexity. |
| Fragment-Trees [65] | Decomposes molecules into fragments organized into acyclic tree structures. | ~100 (structural) | Efficiently handles large, complex molecules; enables parallel processing with Transformers. | Dependent on the rules used for fragmentation. |
Experimental Protocols for Benchmarking Molecular VAEs
To ensure reproducible and comparable results in the field, researchers must adhere to standardized benchmarking protocols. The following section outlines detailed methodologies for evaluating the performance of VAE models using different molecular representations.
Protocol 1: Evaluating Reconstruction and Validity
This protocol assesses a model's ability to accurately encode and decode molecular structures.
- Dataset Curation: Employ a standardized benchmark dataset such as ZINC250K [65] or MOSES [3]. Apply drug-likeness filters (e.g., molecular weight ≤ 500, defined hydrogen bond donors/acceptors) to ensure pharmaceutical relevance [5].
- Data Splitting: Split the dataset into training, validation, and test sets using a standard hold-out method (e.g., 80/10/10 split). Ensure no data leakage between splits.
- Model Training: Train the VAE model (e.g., CVAE, JT-VAE, FRATTVAE) on the training set. The model's objective is to minimize a loss function combining reconstruction error and the Kullback-Leibler (KL) divergence:
L_total = L_recon + β * L_KL, whereβis a weighting coefficient [65]. - Reconstruction Accuracy Calculation: For each molecule in the test set, encode it to the latent space and then decode it. Calculate the reconstruction accuracy as the proportion of test molecules for which the decoded SMILES string matches the original input string exactly [65].
- Validity Calculation: Sample a large number of latent vectors (e.g., 10,000) from the prior distribution (e.g.,
N(0, I)). Decode each vector and use a cheminformatics toolkit like RDKit to check the chemical validity of the generated structure. Validity is the percentage of generated molecules that are chemically valid [20].
Protocol 2: Assessing Conditional Generation
This protocol evaluates a model's capability to generate molecules with specific target properties.
- Conditional Model Setup: Implement a Conditional VAE (CVAE) where a vector of molecular properties (e.g., molecular weight, LogP, TPSA) is concatenated with the input representation and the latent vector [21].
- Property Prediction Head: For non-conditional VAEs, a property predictor can be finetuned from the pretrained encoder. This predictor supplies a conditioning signal applied to the latent prior and decoder [5].
- Conditional Generation: Sample a latent vector
zand concatenate it with a condition vectorccontaining the desired property values. Decode the combined[z, c]to generate novel molecules. - Success Metric Evaluation: Generate a large set of molecules for a given condition
c. Calculate the success rate as the fraction of generated molecules that meet all target properties within a specified tolerance. Analyze the distribution of docking scores or other complex properties to confirm a statistically significant shift towards the desired profile [5].
Protocol 3: Mitigating Posterior Collapse
Posterior collapse, where the model ignores the latent space, is a common failure mode. The following protocol is adapted from the PCF-VAE study [3] [66].
- Input Preprocessing: Convert SMILES strings into a more robust representation like GenSMILES to reduce complexity and long-range dependencies.
- Loss Function Reparameterization: Modify the standard VAE loss function to include additional constraints or a diversity-promoting term that actively penalizes the collapse of the latent space.
- Diversity Layer Integration: Introduce a dedicated "diversity layer" between the latent space and the decoder. This layer uses a tunable diversity parameter to explicitly control the trade-off between the validity and diversity of generated molecules.
- Validation: Monitor the KL divergence term during training. A near-zero value indicates posterior collapse. Evaluate the internal diversity (intDiv) of generated molecules, which for PCF-VAE ranges from 85.87% to 89.01% [3].
Workflow Visualization: From Representation to Generation
The following diagram illustrates the logical and computational workflow for generating molecules using advanced representation learning with VAEs, integrating the concepts from the discussed protocols.
Diagram 1: Molecular Generation Workflow
The Scientist's Toolkit: Essential Research Reagents & Solutions
Successful implementation of the aforementioned protocols requires a suite of specialized software tools and computational resources. The following table details the key "research reagents" for this digital laboratory.
Table 2: Essential Computational Reagents for Molecular VAE Research
| Tool/Resource | Type | Primary Function | Application Example |
|---|---|---|---|
| RDKit [20] [21] | Cheminformatics Library | Checks molecular validity, calculates properties (e.g., LogP, TPSA), and handles canonicalization. | Used in post-generation validation and property calculation for conditional generation. |
| PubChem [5] | Chemical Database | Source of millions of drug-like molecules for large-scale pretraining of foundational models. | Curating a dataset of 79 million compounds for training STAR-VAE. |
| ZINC/MOSES [65] | Benchmark Datasets | Provides standardized training and test sets for reproducible evaluation of generative models. | Benchmarking reconstruction accuracy and novelty against established baselines. |
| SELFIES [5] | Molecular Representation | A string-based representation that guarantees 100% syntactic validity. | Input representation for transformer-based VAEs like STAR-VAE to ensure valid outputs. |
| GenSMILES [63] [3] | Molecular Representation | A SMILES-like representation that reduces complexity and improves validity via derivation rules. | Preprocessing input for PCF-VAE to mitigate posterior collapse and enhance diversity. |
| Low-Rank Adaptation (LoRA) [5] | Fine-Tuning Method | Enables parameter-efficient adaptation of large pretrained models with limited property data. | Fast fine-tuning of a pretrained VAE for a new protein target with few known actives. |
| AbDb/abYbank [67] | Structural Database | A specialized database of immunoglobulin structures for class-specific 3D generation. | Training the Ig-VAE model to generate novel, designable antibody backbones. |
The journey from simple string representations to complex, information-rich 3D structures marks a significant maturation of generative models in chemistry. Framed within the broader thesis of VAE research, this evolution is not merely incremental but foundational, enabling models to capture the intricate rules of chemistry and spatial reality with increasing fidelity. Representations like GenSMILES and SELFIES have largely solved the problem of syntactic validity, while graph-based and fragment-tree approaches natively encode structural constraints. The frontier now lies in the seamless integration of 3D complexity, as exemplified by UAE-3D, which paves the way for generative models that can directly reason about molecular interactions in biological systems. As these representations and the VAEs that leverage them continue to advance, they will undoubtedly accelerate the discovery of novel therapeutics and materials, solidifying their role as an indispensable tool in the modern scientist's computational arsenal.
Generative artificial intelligence (GenAI) has emerged as a transformative tool in molecular sciences, offering the potential to systematically navigate the vast chemical space estimated to contain up to 10^60 drug-like molecules [68] [5]. Among generative architectures, variational autoencoders (VAEs) have established themselves as fundamental building blocks for molecular generation due to their ability to learn smooth, continuous latent representations of chemical structures [69] [5]. However, standalone VAEs often generate overly smooth distributions with limited structural diversity and face challenges in producing novel molecular entities with optimized properties [70] [69].
To overcome these limitations, researchers have developed sophisticated hybrid frameworks that integrate VAEs with other generative paradigms, particularly generative adversarial networks (GANs) and diffusion models. These hybrid approaches leverage the complementary strengths of each architecture: the stable latent space learning of VAEs, the high-fidelity sample generation of GANs, and the precise distribution modeling of diffusion processes [70] [69]. The integration has demonstrated significant improvements in generating chemically valid, structurally diverse, and functionally relevant molecules, thereby accelerating the discovery of novel therapeutic compounds [70] [43].
Key Hybrid Architectures and Performance
VAE-GAN Integration Frameworks
The integration of VAEs and GANs represents a powerful synergy for molecular generation. In this architecture, VAEs provide robust feature extraction and latent space organization, while GANs enhance structural diversity and generative fidelity through adversarial training [70]. The VGAN-DTI framework exemplifies this approach, combining these architectures with multilayer perceptrons (MLPs) for drug-target interaction (DTI) prediction. This model employs a VAE component to encode molecular structures into latent representations using a probabilistic encoder-decoder structure, while the GAN component generates diverse molecular candidates through adversarial training between generator and discriminator networks [70].
The VAE encoder in these frameworks typically processes molecular fingerprint vectors through fully connected layers with ReLU activation, producing mean (μ) and log-variance (log σ²) parameters that define the latent space distribution. The decoder reconstructs molecular structures from latent samples, with the loss function combining reconstruction loss with Kullback-Leibler (KL) divergence to regularize the latent space [70]. Simultaneously, the GAN generator transforms random noise into molecular representations, while the discriminator distinguishes between real and generated compounds, creating an adversarial training dynamic that enhances output quality [70].
Table 1: Performance Comparison of Hybrid Generative Models in Molecular Design
| Model Architecture | Primary Application | Key Metrics | Performance Advantages |
|---|---|---|---|
| VGAN-DTI [70] | Drug-target interaction prediction | Accuracy: 96%, Precision: 95%, Recall: 94%, F1-score: 94% | Outperforms existing methods in prediction accuracy |
| STAR-VAE [5] | Conditional molecular generation | Validity: >90%, Diversity: High, Optimized docking scores | Statistically significant improvement in binding affinities for protein targets |
| ScafVAE [43] | Multi-objective drug design | High QED/SA scores, Strong binding affinity, Optimized ADMET | Effective dual-target drug generation against cancer resistance mechanisms |
| GaUDI (Diffusion) [69] | Inverse molecular design | 100% validity, Multi-objective optimization | Successfully optimizes for single and multiple objectives simultaneously |
VAE-Diffusion Model Hybrids
Diffusion models have recently emerged as competitive alternatives to GANs, employing iterative forward and reverse processes that gradually add and remove noise from data samples [71] [69]. When integrated with VAEs, diffusion models can operate efficiently in the compressed latent spaces learned by VAEs, significantly reducing computational requirements while maintaining high-quality generation [71] [69].
The GaUDI (Guided Diffusion for Inverse Molecular Design) framework exemplifies this approach, combining an equivariant graph neural network for property prediction with a generative diffusion model [69]. This hybrid architecture achieves remarkable 100% validity in generated structures while successfully optimizing for both single and multiple objectives [69]. The VAE component provides a structured latent space where molecular representations are organized according to chemical properties, while the diffusion process enables precise traversal and sampling of this space for targeted molecular generation.
Advanced Transformer-Enhanced VAEs
Recent advancements have incorporated transformer architectures into VAE frameworks to enhance sequence modeling capabilities. STAR-VAE (Selfies-encoded, Transformer-based, AutoRegressive Variational Auto Encoder) represents a scalable latent-variable framework with a transformer encoder and autoregressive transformer decoder [5]. This model is trained on millions of drug-like molecules from PubChem using SELFIES representations to guarantee syntactic validity, and employs a principled conditional latent-variable formulation for property-guided generation [5].
The transformer enhancement allows STAR-VAE to capture long-range dependencies in molecular structures more effectively than traditional recurrent neural networks, while maintaining the beneficial probabilistic framing of VAEs. This architecture supports both unconditional exploration and property-aware steering of molecular generation, demonstrating competitive performance on standard benchmarks including GuacaMol and MOSES [5].
Experimental Protocols and Methodologies
Protocol: Implementing VAE-GAN-DTI Framework
Objective: Establish a hybrid VAE-GAN framework for drug-target interaction prediction with optimized molecular generation capabilities.
Materials and Data Requirements:
- Molecular datasets (e.g., BindingDB, PubChem) with validated drug-target pairs
- Computational resources: GPU clusters with minimum 16GB VRAM
- Software: Python 3.8+, PyTorch/TensorFlow, RDKit, specialized libraries (DeepChem, OGB)
Procedure:
Data Preprocessing:
- Convert molecular structures to standardized representations (SMILES, SELFIES, or molecular graphs)
- Generate molecular fingerprint vectors (ECFP, Morgan fingerprints) for input encoding
- Normalize and split data into training (70%), validation (15%), and test sets (15%)
VAE Component Implementation:
- Configure encoder network with 2-3 hidden layers (512 units each, ReLU activation)
- Implement stochastic latent layer with separate dense layers for μ and log σ² parameters
- Design decoder network mirroring encoder architecture with Bernoulli output distribution
- Define VAE loss function: ℒVAE = 𝔼qθ(z|x)[log pφ(x|z)] - DKL[q_θ(z|x) || p(z)]
GAN Component Integration:
- Implement generator network that transforms latent vectors to molecular representations
- Configure discriminator with leaky ReLU activation and sigmoid output
- Define adversarial losses:
- Discriminator loss: ℒD = 𝔼z∼pdata(x)[log D(x)] + 𝔼z∼pz(z)[log(1 - D(G(z)))]
- Generator loss: ℒG = -𝔼z∼pz(z)[log D(G(z))]
Multitask Training Protocol:
- Phase 1: Pre-train VAE component using reconstruction and KL loss
- Phase 2: Jointly train VAE-GAN with combined objective function
- Phase 3: Fine-tune with property prediction heads for target-specific optimization
- Training parameters: Adam optimizer (lr=0.001), batch size=128, 500-1000 epochs
Validation and Evaluation:
- Assess reconstruction accuracy and latent space organization
- Evaluate generative quality through validity, uniqueness, and novelty metrics
- Validate DTI predictions using accuracy, precision, recall, and F1-score
- Conduct ablation studies to verify component contributions [70]
Protocol: Conditional Molecular Generation with STAR-VAE
Objective: Implement property-guided molecular generation using transformer-enhanced VAE architecture.
Procedure:
Molecular Representation:
- Convert molecules to SELFIES representations to ensure syntactic validity
- Implement byte-pair encoding (BPE) tokenization for sequence processing
Transformer Architecture Configuration:
- Design bi-directional transformer encoder for latent space inference
- Implement autoregressive transformer decoder for sequence generation
- Configure model dimensions: latent dimension=512, transformer layers=8, attention heads=8
Conditional Generation Mechanism:
- Train property prediction heads on latent representations
- Integrate conditioning signal throughout architecture: prior p(z|c), inference network q(z|x,c), and decoder p(x|z,c)
- Implement low-rank adaptation (LoRA) for parameter-efficient fine-tuning
Training Protocol:
- Pre-training: Train on large-scale molecular dataset (e.g., 79M PubChem compounds)
- Property prediction: Train surrogate models on labeled data
- Conditional fine-tuning: Adapt model to specific property objectives with limited data
- Employ gradient clipping, learning rate scheduling, and early stopping [5]
Protocol: Multi-Objective Optimization with ScafVAE
Objective: Generate molecules satisfying multiple property constraints using scaffold-aware VAE framework.
Procedure:
Scaffold-Based Molecular Encoding:
- Implement perplexity-inspired fragmentation to identify optimal bond breakpoints
- Calculate bond perplexity using pre-trained masked graph model
- Encode molecular graphs using sequential fragmentation approach
Bond Scaffold Generation:
- Generate bond scaffolds (fragments without specified atom types) using recurrent graph neural network
- Assemble scaffolds through iterative bond connection
- Decorate scaffolds with atom types to form complete molecules
Multi-Objective Optimization:
- Train surrogate models for diverse property predictions (binding affinity, ADMET, QED, SA)
- Implement Bayesian optimization in latent space for property targeting
- Configure multi-objective reward function with weighted property combinations
Evaluation Framework:
- Assess multi-property satisfaction rates
- Validate binding interactions through molecular docking simulations
- Confirm binding stability with molecular dynamics simulations [43]
Table 2: Research Reagent Solutions for Hybrid Model Implementation
| Reagent/Resource | Type | Function | Implementation Example |
|---|---|---|---|
| SELFIES [5] | Molecular Representation | Guarantees 100% syntactic validity in generated structures | STAR-VAE uses SELFIES to eliminate invalid molecular strings |
| BindingDB [70] | Dataset | Provides drug-target interaction data for training | VGAN-DTI training and validation using curated BindingDB entries |
| PubChem [5] | Dataset | Large-scale molecular database for pretraining | 79M drug-like molecules for foundation model training |
| Low-Rank Adaptation (LoRA) [5] | Optimization Technique | Enables parameter-efficient fine-tuning with limited data | STAR-VAE adaptation to new properties with minimal parameters |
| Bayesian Optimization [69] | Optimization Method | Efficiently navigates high-dimensional latent spaces | Identifies optimal latent vectors for multi-property satisfaction |
| Graph Neural Networks [43] | Architecture Component | Processes molecular graph representations | ScafVAE encoder and decoder for graph-based generation |
| Molecular Fingerprints [70] | Feature Representation | Encodes molecular structures as numerical vectors | Input features for VAE encoder in hybrid frameworks |
Visualization of Molecular Optimization Workflow
The integration of VAEs with GANs and diffusion models represents a significant advancement in generative molecular design, effectively addressing limitations of individual architectures while leveraging their complementary strengths. These hybrid frameworks demonstrate enhanced capability to generate chemically valid, structurally diverse, and functionally optimized molecules, as evidenced by their performance in rigorous benchmarks and prospective applications [70] [5] [43].
The protocols and architectures detailed in this work provide researchers with practical frameworks for implementing these advanced generative approaches. As the field evolves, future developments will likely focus on improving model interpretability, expanding conditional control capabilities, and enhancing integration with experimental validation pipelines. Through continued refinement and application, hybrid generative models stand to substantially accelerate the discovery and optimization of novel therapeutic compounds.
Benchmarking VAE Performance: Metrics, Platforms, and Comparative Analysis
Standardized Benchmarking with the MOSES Platform
The discovery of novel molecular structures for pharmaceutical applications represents a significant challenge due to the vastness of chemical space, which is estimated to contain between 10²³ and 10⁸⁰ pharmacologically sensible compounds [72] [73]. Deep generative models, particularly Variational Autoencoders (VAEs), have emerged as powerful tools for de novo molecular design, offering the potential to efficiently explore this extensive chemical space [74]. However, the rapid evolution of these models has created a critical need for standardized evaluation protocols to ensure fair comparison and validate advancements [74] [75].
The Molecular Sets (MOSES) benchmarking platform was developed to address this need by providing a unified framework for training, evaluating, and comparing molecular generative models [76] [72]. As VAE-based approaches continue to evolve—addressing challenges such as posterior collapse and synthetic accessibility [3]—MOSES serves as an essential tool for quantifying their performance and tracking progress in the field. This application note provides a comprehensive guide to utilizing MOSES within VAE research, detailing its core components, evaluation metrics, and experimental protocols.
The MOSES Platform: Core Components
Standardized Datasets and Data Processing
MOSES provides a curated dataset derived from the ZINC Clean Leads collection, ensuring consistency across research efforts [76] [72]. The dataset undergoes rigorous filtering and preprocessing to maximize its relevance for early-stage drug discovery.
Key Dataset Characteristics:
- Source: ZINC Clean Leads collection [76]
- Size: 1,936,962 molecular structures after filtering [76]
- Standard Splits: Training set (~1.6M molecules), test set (176k molecules), and scaffold test set (176k molecules) [76]
- Molecular Weight Range: 250-350 Daltons [76]
- Filters Applied: Medicinal chemistry filters (MCFs), PAINS filters, and restrictions on rotatable bonds (≤7), XlogP (≤3.5), and atom types [76]
The scaffold test set is particularly valuable for VAE research, as it contains unique Bemis-Murcko scaffolds not present in the training set, enabling researchers to assess how well models generalize and generate novel molecular frameworks [76].
Molecular Representations
MOSES supports various molecular representations, each with distinct advantages for VAE-based generation:
- SMILES (Simplified Molecular-Input Line-Entry System): Linear string representations that are compatible with natural language processing architectures but prone to syntactic invalidity if not properly constrained [72] [73].
- SELFIES (Self-Referencing Embedded Strings): A grammar-based representation that guarantees 100% syntactic validity, making it increasingly popular for VAE implementations [5] [77].
- Graph Representations: Direct encoding of molecular structures as atoms (nodes) and bonds (edges), which naturally capture molecular topology but require specialized architectures [72] [73].
Evaluation Metrics
MOSES implements a comprehensive suite of metrics to evaluate generated molecules across multiple dimensions [76] [72]:
Table 1: Key Evaluation Metrics in MOSES
| Metric | Description | Interpretation |
|---|---|---|
| Validity | Fraction of generated strings that correspond to valid molecules | Higher values indicate better model performance at generating chemically plausible structures |
| Uniqueness | Proportion of valid molecules that are distinct from one another | Measures diversity and prevents mode collapse |
| Novelty | Fraction of unique valid molecules not present in the training set | Assesses ability to generate new structures rather than memorizing training data |
| Fréchet ChemNet Distance (FCD) | Distance between distributions of generated and test set molecules in the chemical/biological activity space | Lower values indicate better alignment with the chemical and biological properties of real molecules |
| Internal Diversity (IntDiv) | Average pairwise similarity between generated molecules | Measures structural variety within the generated set |
| Scaffold Similarity (Scaff) | Cosine similarity between vectors of scaffold frequencies in generated and test sets | Assesses whether the model captures the distribution of core molecular frameworks |
| Filters | Proportion of molecules passing medicinal chemistry filters | Estimates practical utility and drug-likeness |
Benchmarking VAE Performance: Protocols and Baseline Results
Standardized Evaluation Protocol
To ensure comparable results across different VAE architectures, MOSES establishes a standardized evaluation workflow:
- Training: Train the VAE model on the official MOSES training set (~1.6 million molecules) [76].
- Generation: Use the trained model to generate 30,000 molecules [72].
- Validation: Filter valid molecules from the generated set using RDKit [76].
- Evaluation: Compute all metrics using only valid molecules, comparing against the MOSES test set [76].
Baseline VAE Performance
MOSES provides benchmark results for several generative models, including VAE architectures. The table below summarizes performance metrics for key models as reported in the MOSES benchmark:
Table 2: Benchmark Performance of Selected Models on MOSES [76]
| Model | Valid (↑) | Unique@10k (↑) | FCD (↓) | Novelty (↑) | IntDiv (↑) |
|---|---|---|---|---|---|
| VAE | 97.67% | 99.84% | 0.099 | 69.49% | 0.9386 |
| JTN-VAE | 100% | 99.96% | 0.395 | 91.43% | 0.8964 |
| AAE | 93.68% | 99.73% | 0.556 | 79.31% | 0.9022 |
| LatentGAN | 89.66% | 99.68% | 0.297 | 94.98% | 0.8867 |
| CharRNN | 97.48% | 99.94% | 0.073 | 84.19% | 0.9242 |
These benchmarks reveal important trade-offs in VAE design. While standard VAEs achieve high validity and uniqueness, they sometimes lag in novelty compared to other architectures, highlighting the challenge of posterior collapse where the model fails to fully utilize the latent space [3].
Advanced VAE Architectures in MOSES Framework
Recent VAE variants have demonstrated improved performance on MOSES metrics by addressing specific limitations:
- PCF-VAE: Specifically designed to mitigate posterior collapse, this architecture achieves 98.01% validity while maintaining 93.77% novelty and 100% uniqueness at diversity level D=1 [3].
- NP-VAE: A graph-based VAE that handles large molecular structures with 3D complexity, showing higher reconstruction accuracy compared to previous models [20].
- STAR-VAE: Incorporates Transformer architecture with SELFIES representation, matching or exceeding baseline performance while enabling property-guided generation through latent space conditioning [5].
Experimental Protocols for VAE Evaluation
Workflow for Benchmarking VAE Models
The following diagram illustrates the complete experimental workflow for evaluating VAE models using the MOSES platform:
Protocol 1: Standard VAE Benchmarking
Objective: Evaluate VAE performance against standard MOSES baselines [76] [72].
Materials:
- MOSES dataset (training, test, and scaffold test sets)
- Computational environment with RDKit installed
- VAE implementation (e.g., PyTorch or TensorFlow)
Procedure:
- Data Preparation:
- Download the MOSES dataset through the official package (
pip install molsets) - Preprocess molecules according to MOSES standards
- Split data into training, test, and scaffold test sets
- Download the MOSES dataset through the official package (
Model Configuration:
- Select molecular representation (SMILES, SELFIES, or graph-based)
- Define encoder-decoder architecture appropriate for the representation
- Set hyperparameters (latent dimension, learning rate, batch size)
Training:
- Train VAE on the MOSES training set (1.6M molecules)
- Monitor reconstruction loss and regularization terms
- Save model checkpoints for generation phase
Generation:
- Generate 30,000 molecules from the trained model
- For latent space models, sample from prior distribution N(0,I)
Evaluation:
- Filter valid molecules using RDKit
- Compute all MOSES metrics on valid molecules
- Compare results against published baselines
Protocol 2: Scaffold Novelty Assessment
Objective: Specifically evaluate the model's ability to generate novel molecular scaffolds [76].
Procedure:
- Follow standard benchmarking protocol through generation phase
- Extract Bemis-Murcko scaffolds from generated molecules
- Compare against scaffolds in training set
- Calculate scaffold novelty as proportion of generated scaffolds not present in training
- Compute scaffold similarity (Scaff) metric between generated and test sets
Protocol 3: Conditional VAE Evaluation
Objective: Assess property-controlled generation using conditional VAE architectures [5].
Materials:
- Property labels (e.g., QED, logP, synthetic accessibility)
- Conditional VAE implementation (e.g., STAR-VAE [5])
Procedure:
- Train conditional VAE with property conditioning
- Generate molecules targeting specific property values
- Evaluate both distribution learning metrics and property optimization success
- Compare with unconditional generation to assess improvement
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Computational Tools for MOSES Benchmarking
| Tool/Resource | Function | Application in VAE Research |
|---|---|---|
| RDKit | Cheminformatics toolkit | Molecular validity checking, descriptor calculation, and visualization |
| PyTorch/TensorFlow | Deep learning frameworks | VAE implementation and training |
| MOSES Python Package | Benchmarking utilities | Standardized dataset loading and metric computation |
| SELFIES Library | Molecular representation | Grammar-based representation guaranteeing 100% validity |
| JTN-VAE Implementation | Graph-based VAE | Baseline for scaffold-based molecular generation |
| ChemNet | Pretrained biological activity predictor | FCD metric computation for chemical/biological distribution comparison |
Analysis of VAE Performance and Research Directions
Interpreting VAE Performance on MOSES
The benchmarking results reveal several key patterns in VAE performance:
- Validity Challenge: Standard SMILES-based VAEs typically achieve 85-98% validity rates [76], while graph-based approaches (e.g., JTN-VAE) and SELFIES-based models can reach near-perfect validity [5].
- Novelty-Diversity Tradeoff: VAEs sometimes struggle with novelty compared to other architectures (e.g., LatentGAN), indicating posterior collapse issues where the model reproduces training examples rather than generating novel structures [3].
- Representation Impact: The choice of molecular representation significantly affects all metrics. SELFIES representations guarantee validity but may require architectural adjustments [5], while graph-based representations naturally capture molecular topology but increase computational complexity [20].
Advanced Analysis: Latent Space Characterization
Beyond standard MOSES metrics, researchers can perform additional latent space analysis:
- Smoothness Assessment: Interpolate between latent points and decode to molecules, evaluating whether intermediate points correspond to valid structures [20].
- Property Correlation: Train property predictors on latent representations to assess whether the space captures chemically meaningful directions [5].
- Cluster Analysis: Project latent space to 2D (e.g., using UMAP or t-SNE) and color by molecular properties to visualize organization.
Future Directions in VAE Benchmarking
Current research trends identified through MOSES benchmarking include:
- Hybrid Models: Combining VAEs with normalizing flows for improved latent space modeling [77].
- 3D-Aware Generation: Extending VAEs to capture stereochemistry and conformational complexity [20].
- Large-Scale Pretraining: Training transformer-based VAEs on millions of compounds followed by fine-tuning [5].
- Multi-Objective Optimization: Developing VAEs that simultaneously optimize multiple chemical properties while maintaining diversity [3] [5].
The MOSES benchmarking platform provides an essential foundation for rigorous evaluation of VAE-based molecular generative models. By offering standardized datasets, comprehensive metrics, and baseline implementations, it enables meaningful comparison across architectures and tracks progress in the field. The protocols outlined in this application note offer researchers a clear pathway for evaluating their VAE implementations, while the analysis of current performance highlights both strengths and limitations of existing approaches. As VAE methodologies continue to evolve, MOSES will play a crucial role in validating advancements and guiding the development of more effective generative models for drug discovery.
In the field of de novo molecular design, generative models, particularly Variational Autoencoders (VAEs), have emerged as powerful tools for exploring the vast chemical space. These models aim to produce novel molecular structures with desirable pharmacological properties, thereby accelerating the drug discovery pipeline. The performance of these generative models is quantitatively assessed using four critical metrics: validity, uniqueness, novelty, and diversity. Validity ensures the generated molecular structures are chemically plausible; uniqueness prevents model redundancy by measuring the generation of duplicate structures; novelty assesses the model's capacity to produce structures not present in the training data; and diversity gauges the chemical variety within the generated set, ensuring a broad exploration of chemical space. This protocol details the application of these metrics for evaluating VAE-based molecular generators, providing researchers with standardized methodologies for model assessment and comparison.
Performance Metrics and Quantitative Comparison
The table below summarizes the performance of several state-of-the-art VAE models based on the key metrics, providing a benchmark for comparison. The metrics are typically evaluated on standard benchmarks like MOSES.
Table 1: Performance Comparison of VAE Models for Molecule Generation
| Model Name | Core Innovation | Validity (%) | Uniqueness (%) | Novelty (%) | Diversity (IntDiv/IntDiv2, %) | Key Application Context |
|---|---|---|---|---|---|---|
| PCF-VAE [3] | Mitigates posterior collapse; uses GenSMILES & diversity layer. | 95.01 - 98.01 | ~100 | 93.77 - 95.01 | 85.87 - 89.01 / 85.87 - 86.33 | De novo drug design |
| TGVAE [4] | Combines Transformer, GNN, and VAE; uses molecular graphs as input. | High (Specific values not reported) | High (Specific values not reported) | High (Specific values not reported) | High (Specific values not reported) | Generative molecular design |
| RGCVAE [78] | Relational Graph Isomorphism Network for encoding; considers stereochemistry. | High (Specific values not reported) | High (Specific values not reported) | High (Specific values not reported) | High (Specific values not reported) | Molecule design & optimization |
| SmilesGEN [44] | Dual-channel VAE integrating SMILES and phenotypic profiles. | High (Specific values not reported) | High (Specific values not reported) | High (Specific values not reported) | High (Specific values not reported) | Phenotypic profile-informed molecule generation |
Experimental Protocols
Protocol 1: Standardized Model Evaluation using the MOSES Benchmark
This protocol provides a step-by-step guide for evaluating a VAE model's performance on the standard MOSES (Molecular Sets) benchmark, ensuring comparable and reproducible results.
A. Prerequisite Setup
- Data Acquisition: Download the MOSES benchmark dataset, which includes standardized training, test, and scaffold-test sets of molecules, typically derived from the ZINC database.
- Model Training: Train your VAE model (e.g., PCF-VAE, TGVAE) on the provided MOSES training set.
- Sample Generation: Use the trained model to generate a large set of molecules (e.g., 30,000) for evaluation.
B. Metric Calculation Workflow The following diagram illustrates the sequential workflow for calculating the four key performance metrics.
C. Step-by-Step Procedure
- Validity Assessment:
- Action: Pass each generated string (e.g., SMILES, GenSMILES) through a chemistry toolkit (e.g., RDKit) to check if it corresponds to a chemically valid molecule.
- Calculation: Validity = (Number of Valid Molecules / Total Number of Generated Molecules) × 100%.
Uniqueness Assessment:
- Action: Remove duplicate molecular structures from the set of valid molecules. Duplicates are identified by canonicalizing the SMILES strings.
- Calculation: Uniqueness = (Number of Unique Valid Molecules / Number of Valid Molecules) × 100%.
Novelty Assessment:
- Action: Check the set of unique, valid molecules against the training set used for the VAE model.
- Calculation: Novelty = (Number of Molecules not in Training Set / Number of Unique Valid Molecules) × 100%.
Diversity Assessment:
- Action: Calculate the internal diversity of the unique, valid, and novel set of molecules. This is often computed as the average Tanimoto similarity (1 - similarity) between all pairs of molecular fingerprints in the set.
- Calculation: Internal Diversity (IntDiv) = 1 - ( 2 / (N*(N-1)) ) × Σ Simᵢⱼ, where N is the set size and Simᵢⱼ is the Tanimoto similarity between molecules i and j. A lower average similarity indicates higher diversity.
Protocol 2: Mitigating Posterior Collapse in VAEs for Improved Diversity
Posterior collapse is a common issue in VAEs where the decoder ignores the latent variables, leading to low diversity in generated samples. This protocol outlines the method used by PCF-VAE to mitigate this problem [3].
A. Objective To reparameterize the VAE loss function and modify the input representation to reduce posterior collapse, thereby enhancing the quality and diversity of generated molecules.
B. Procedure
- Input Representation Transformation:
- Convert standard SMILES strings into GenSMILES representations. This process is designed to preserve semantic information while reducing the complexity of the string representation, making it easier for the model to learn meaningful patterns [3].
- Optionally, incorporate key molecular properties (e.g., molecular weight, LogP, TPSA) directly into the GenSMILES representation to guide the generation towards molecules with desired properties [3].
Loss Function Reparameterization:
- Modify the standard VAE loss function, which consists of a reconstruction loss and the Kullback-Leibler (KL) divergence term. The specific reparameterization strategy used by PCF-VAE is tailored to prevent the KL term from vanishing, which is a hallmark of posterior collapse [3].
Incorporation of a Diversity Layer:
- Introduce a dedicated layer between the latent space and the decoder. This layer is designed to explicitly control the diversity of the generated outputs, allowing for a more granular manipulation of the variety in the generated molecular set [3].
Table 2: Key Research Reagents and Computational Tools
| Item Name | Function / Purpose | Application Example in Protocol |
|---|---|---|
| ZINC / MOSES Dataset | A curated, publicly available database of commercially available drug-like molecules, often used as a standard benchmark. | Used as the training and benchmark data in Protocol 1 for consistent model evaluation [78]. |
| RDKit | An open-source cheminformatics toolkit used for manipulating and analyzing chemical structures. | Essential for validity checks (canonicalizing SMILES, checking chemical rules) and calculating molecular fingerprints for diversity metrics in Protocol 1 [3]. |
| GenSMILES | A transformed version of SMILES strings designed to reduce complexity while preserving molecular semantics. | Used in PCF-VAE (Protocol 2) as a more robust input representation to facilitate model learning and improve validity [3]. |
| Tanimoto Similarity | A metric calculated from molecular fingerprints (e.g., Morgan fingerprints) to quantify the structural similarity between two molecules. | The core calculation for determining the internal diversity (IntDiv) of the generated set in Protocol 1 [3]. |
| Graph Neural Network (GNN) | A type of neural network that operates directly on graph structures, naturally representing molecules (atoms as nodes, bonds as edges). | Employed in models like TGVAE and RGCVAE to capture complex structural relationships more effectively than string-based representations [4] [78]. |
| Hierarchical Prior (ARD) | A statistical prior used in the latent space to automatically identify and prune away irrelevant latent dimensions. | Used in ARD-VAE to find the relevant latent dimensions without empirical tuning, improving model efficiency and interpretation [79] [80]. |
Model Architecture and Evaluation Workflow
The following diagram integrates the architectural innovations of advanced VAEs with the standardized evaluation protocol, providing a complete overview from molecule generation to performance assessment.
Comparative Analysis of State-of-the-Art VAE Models
Variational Autoencoders (VAEs) have emerged as a foundational architecture in generative modeling, particularly for structured data like molecular graphs and 3D shapes. Within drug discovery, VAEs offer a principled framework for learning smooth, interpretable latent spaces of molecular structures, enabling efficient exploration and optimization of novel compounds with desirable properties. This analysis provides a comparative examination of contemporary VAE architectures—TGVAE, STAR-VAE, and LoG3D—focusing on their architectural innovations, performance benchmarks, and practical applications in molecular generation research. The content is structured to serve as a technical reference for researchers and scientists engaged in AI-driven drug development, detailing experimental protocols, key reagents, and analytical workflows specific to this domain.
Comparative Analysis of State-of-the-Art VAE Architectures
The table below summarizes the core attributes, strengths, and applications of three leading VAE models.
Table 1: Comparison of State-of-the-Art VAE Models
| Model Name | Core Architectural Innovation | Primary Application Domain | Key Advantages | Quantitative Performance Highlights |
|---|---|---|---|---|
| TGVAE (Transformer Graph VAE) [4] [81] | Integrates Transformer, Graph Neural Network (GNN), and VAE components. [4] | Generative molecular design for drug discovery [4] [81] | Effectively captures complex structural relationships within molecules; generates diverse and novel structures [4] [81] | Outperforms existing approaches; generates a larger collection of diverse molecules [4] [81] |
| STAR-VAE [5] | Employs a bi-directional Transformer encoder and an autoregressive Transformer decoder; uses SELFIES representation. [5] | Scalable and controllable molecular generation [5] | Guarantees syntactic validity; enables property-guided conditional generation; supports parameter-efficient fine-tuning [5] | Matches or exceeds baselines on GuacaMol and MOSES benchmarks; shows improved docking score distributions [5] |
| LoG3D [82] | A 3D VAE using Unsigned Distance Fields (UDFs) and a local-to-global (LoG) architecture with 3D convolutions and sparse transformers. [82] | Ultra-high-resolution 3D shape modeling [82] | Naturally handles complex, non-manifold geometries; scales to unprecedented resolutions (up to 2048³) [82] | Achieves state-of-the-art reconstruction accuracy and generative quality with smoother surfaces [82] |
Detailed Experimental Protocols
Protocol for Training and Evaluating TGVAE
Objective: To train and evaluate the Transformer Graph VAE (TGVAE) for generating novel, diverse, and chemically valid molecular structures.
Materials:
- Hardware: High-performance computing cluster with modern GPUs (e.g., NVIDIA H100 or A100).
- Software: Python 3.8+, PyTorch or TensorFlow, deep graph learning libraries (e.g., PyTorch Geometric), RDKit for cheminformatics.
- Data: Public molecular datasets such as ZINC15 or ChEMBL, preprocessed into molecular graph representations (atoms as nodes, bonds as edges).
Procedure:
- Data Preprocessing:
- Convert molecular SMILES strings from the dataset into graph representations using RDKit.
- Node features: Encode atom type, degree, hybridization, etc.
- Edge features: Encode bond type, conjugation, etc.
- Split the dataset into training, validation, and test sets (e.g., 80/10/10).
Model Training:
- Initialization: Initialize TGVAE components: GNN encoder, Transformer layers, and decoder.
- Loss Function: Optimize the combined objective:
- Reconstruction Loss: Cross-entropy loss for graph reconstruction.
- KL Divergence: Regularization term to enforce a structured latent space,
p(𝐳). Techniques to mitigate posterior collapse are employed [83]. Loss = Reconstruction Loss + β * KL Divergence
- Training Loop: Train for a predetermined number of epochs (e.g., 500) using an optimizer like Adam. Monitor loss on the validation set for early stopping.
Model Evaluation:
- Generative Performance: Sample latent vectors
zfrom the priorp(𝐳)and decode them into new molecular graphs [83]. - Metrics:
- Validity: Percentage of generated graphs that form chemically valid molecules.
- Uniqueness: Percentage of unique molecules among valid ones.
- Diversity: Measures of structural diversity among generated molecules.
- Novelty: Fraction of generated molecules not present in the training data.
- Comparative Benchmarking: Compare these metrics against existing molecular generation models.
- Generative Performance: Sample latent vectors
Protocol for Conditional Generation with STAR-VAE
Objective: To perform property-guided molecular generation using the conditional formulation of STAR-VAE.
Materials:
- Model: Pretrained STAR-VAE model [5].
- Data: A smaller dataset of molecules annotated with the target property (e.g., docking scores from the Tartarus benchmark) [5].
Procedure:
- Property Predictor Training:
- Attach a property prediction head (a small neural network) to the frozen encoder of the pretrained STAR-VAE.
- Train this predictor on the labeled dataset to predict molecular properties from latent codes.
Conditional Finetuning with LoRA:
- Integrate Low-Rank Adaptation (LoRA) modules into both the encoder and decoder of STAR-VAE [5].
- The conditioning signal from the property predictor is applied to the latent prior, the inference network, and the decoder [5].
- Finetune only the LoRA parameters and the property predictor on the target property data. This leverages the pretrained knowledge while efficiently adapting to the new task.
Conditional Generation & Evaluation:
- For generation, sample a latent vector
zand condition it on a desired property value. - Decode the conditioned latent vector to generate a SELFIES string, which is guaranteed to be syntactically valid [5].
- Evaluation: Generate a large set of molecules and analyze the distribution of the target property (e.g., docking score). Successful conditioning is indicated by a statistically significant shift in the distribution towards more desirable values compared to unconditional generation [5].
- For generation, sample a latent vector
Architectural Workflows and Signaling Pathways
The following diagrams illustrate the core architectures and processes of the featured VAE models.
TGVAE Molecular Graph Encoding
This diagram visualizes the process of encoding a molecular graph into a latent representation using TGVAE.
STAR-VAE Conditional Generation
This diagram outlines the conditional generation workflow of STAR-VAE, incorporating property prediction and LoRA fine-tuning.
LoG3D Local-to-Global 3D Shape Modeling
This diagram depicts the local-to-global partitioning and processing strategy of the LoG3D model for high-resolution 3D shape modeling.
The Scientist's Toolkit: Key Research Reagents & Materials
This section catalogs essential computational "reagents" and resources for implementing and experimenting with state-of-the-art VAEs in molecular research.
Table 2: Essential Research Reagents and Computational Tools
| Item Name | Function/Description | Relevance to VAE Experiments |
|---|---|---|
| SELFIES Representations [5] | A string-based molecular representation that guarantees 100% syntactic validity. | Used as input to models like STAR-VAE to ensure all generated strings correspond to valid molecules, overcoming limitations of SMILES [5]. |
| Molecular Graphs | Representation of molecules as graphs (atoms as nodes, bonds as edges). | Serves as the direct input for graph-based models like TGVAE, enabling them to capture complex structural relationships more effectively than string models [4] [81]. |
| Unsigned Distance Fields (UDFs) [82] | A 3D shape representation that defines the distance to the nearest surface without a sign, naturally handling open and non-manifold geometries. | Core representation for LoG3D-VAE, providing topological flexibility and robustness for 3D shape modeling without requiring watertight meshes [82]. |
| Low-Rank Adaptation (LoRA) [5] | A parameter-efficient fine-tuning technique that updates a small set of parameters by injecting trainable rank decomposition matrices into model layers. | Used in STAR-VAE to enable fast adaptation and conditional fine-tuning with limited property data, avoiding the cost of full model retraining [5]. |
| Graph Neural Networks (GNNs) | A class of neural networks designed to operate on graph-structured data. | A core component of TGVAE's encoder, responsible for aggregating information from a molecule's local atomic environment to build meaningful node and graph-level embeddings [4] [81]. |
Reconstruction Accuracy and Generative Performance Evaluation
Evaluating the performance of variational autoencoders (VAEs) for molecular generation is a critical step in developing reliable in-silico drug design tools. This document provides a standardized framework for assessing two core capabilities: reconstruction accuracy, which measures a model's ability to faithfully encode and decode molecular structures, and generative performance, which evaluates the quality, diversity, and utility of novel generated molecules. The protocols outlined herein are designed for researchers developing and applying molecular VAEs, enabling comparable and reproducible benchmarking across different models and datasets.
Core Evaluation Metrics and Quantitative Benchmarks
A robust evaluation requires multiple metrics to capture different aspects of model performance. The following metrics are essential for a comprehensive assessment.
Table 1: Key Metrics for Evaluating Molecular VAEs
| Metric Category | Specific Metric | Definition and Purpose | Reported Performance (Model) |
|---|---|---|---|
| Reconstruction | Reconstruction Accuracy (Recon) | Proportion of test-set molecules perfectly reconstructed from their latent vector [65]. | 100% validity (NP-VAE) [20] |
| Validity & Uniqueness | Validity | Percentage of generated molecules that are chemically valid [3]. | 98.01% (PCF-VAE) [3] |
| Uniqueness | Percentage of non-duplicate molecules among the valid generated structures [3]. | 100% (PCF-VAE) [3] | |
| Diversity | Internal Diversity (intDiv) | Measures structural variety within a set of generated molecules [3]. | 85.87-89.01% (PCF-VAE) [3] |
| Distribution Learning | Fréchet ChemNet Distance (FCD) | Measures similarity between the distributions of generated and test-set molecules [65]. | High accuracy across datasets (FRATTVAE) [65] |
| Novelty | Novelty | Proportion of generated molecules not present in the training data [3]. | 93.77-95.01% (PCF-VAE) [3] |
Standardized Experimental Protocols
Protocol 1: Evaluating Reconstruction Accuracy
This protocol assesses a VAE's ability to learn a lossless mapping between molecular structure and latent space.
- Data Splitting: Start with a curated dataset (e.g., ZINC250K, MOSES). Split it into training (∼80%), validation (∼10%), and test (∼10%) sets, ensuring no data leakage [20] [65].
- Model Training: Train the VAE model on the training set. Use the validation set for hyperparameter tuning and to determine early stopping points.
- Reconstruction:
- For each molecule in the test set, encode it to its latent representation
z. - Decode
zto generate a new moleculeM'.
- For each molecule in the test set, encode it to its latent representation
- Accuracy Calculation:
- Compare the SMILES string or molecular graph of
M'with the original input moleculeM. - Calculate Reconstruction Accuracy as the percentage of test molecules for which
M'is identical toM[65]. - For a more robust estimate, some studies use a Monte Carlo approach, performing multiple encodings and decodings per test molecule [20].
- Compare the SMILES string or molecular graph of
Protocol 2: Benchmarking Generative Performance
This protocol evaluates the model's performance as a generator of novel, valid, and diverse molecules.
- Unconditional Sampling: Sample a large number (e.g., 10,000) of latent vectors
zfrom the prior distribution (typically a standard normal distribution,N(0, I)). - Decoding: Decode each sampled
zinto a molecule. - Post-generation Analysis:
- Validity Check: Use a tool like RDKit to check the chemical validity of each generated molecule and calculate the validity rate [20] [3].
- Uniqueness & Novelty: Remove duplicates to calculate uniqueness. Check the remaining molecules against the training set to calculate novelty.
- Diversity & Distribution Metrics: Calculate internal diversity (intDiv) and FCD for the set of valid, unique generated molecules [3] [65].
Protocol 3: Assessing Conditional Generation
This protocol tests the model's ability to generate molecules with targeted properties.
- Model Training: Train a Conditional VAE (CVAE) or a property predictor on the VAE's latent space using property-annotated data (e.g., logP, molecular weight, binding affinity) [5] [84].
- Conditional Sampling:
- For CVAEs, provide a target property value
cduring the sampling process. - For latent space optimization, use an optimization algorithm (e.g., Bayesian optimization) to find regions in the latent space that maximize the predicted property value [55].
- For CVAEs, provide a target property value
- Validation:
- Generate molecules from the conditioned latent vectors.
- Validate that the generated molecules possess the target property by using the predictor model or, ideally, more accurate simulation/experimental methods.
Experimental Workflow Visualization
The following diagram illustrates the logical flow of the core evaluation protocols.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for Molecular VAE Evaluation
| Tool Category | Specific Tool / Resource | Function in Evaluation |
|---|---|---|
| Benchmark Datasets | ZINC250K [65], MOSES [3] [65], GuacaMol [5] [65] | Standardized, curated sets of drug-like molecules for training and benchmarking model performance. |
| Natural Product Libraries | SuperNatural II [65], DrugBank [20] | Used to test model performance on large, complex molecular structures with 3D chirality [20] [65]. |
| Molecular Representation | SMILES [3] [84], SELFIES [5], Molecular Graphs [4] [43] | Different input representations for VAEs, each with trade-offs between validity, ease of use, and structural explicitness. |
| Cheminformatics Toolkit | RDKit [20] [55] | Open-source toolkit essential for processing molecules, checking validity, calculating descriptors, and generating fingerprints. |
| Evaluation Benchmarks | MOSES Benchmark [5] [3], GuacaMol Benchmark [5] | Provide standardized scoring protocols and metrics to ensure fair and consistent model comparisons. |
| Property Prediction | Molecular Docking (e.g., Tartarus [5]), ADMET predictors [43] | Used to validate the functional utility and binding affinity of generated molecules in conditional generation tasks. |
Assessing Chemical Space Exploration and Scaffold Diversity
The exploration of chemical space and the generation of diverse molecular scaffolds are fundamental challenges in AI-driven drug discovery. The vastness of the drug-like chemical space, estimated at 10^23 to 10^60 molecules, makes exhaustive exploration impossible [3] [21]. Variational Autoencoders (VAEs) have emerged as a powerful generative modeling approach, projecting discrete molecular structures into a continuous, lower-dimensional latent space where optimization and exploration become tractable [20] [53]. A key challenge lies in evaluating how effectively these models explore this space and generate novel, diverse, and valid scaffolds–the core molecular frameworks that define compound families and are crucial for scaffold hopping in lead optimization [19] [85]. This Application Note provides a structured framework for quantitatively assessing the chemical space exploration and scaffold diversity of molecular VAEs, featuring standardized metrics, detailed experimental protocols, and a curated toolkit for researchers.
Quantitative Assessment of VAE Performance
A comprehensive assessment of a VAE's generative capabilities requires evaluating multiple interdependent performance criteria. The metrics below provide a quantitative foundation for model benchmarking.
Table 1: Key Quantitative Metrics for Assessing VAE-Generated Molecules
| Metric Category | Specific Metric | Description and Computational Method | Interpretation and Ideal Value |
|---|---|---|---|
| Chemical Validity | Validity Rate | Percentage of generated SMILES/SELFIES strings that correspond to a chemically valid molecule (e.g., checked via RDKit). | Higher is better. SELFIES-based models often achieve ~100% [5]. |
| Uniqueness | Uniqueness Rate | Percentage of valid molecules that are duplicates within a generated set (e.g., 10,000 molecules). | High rate indicates model avoids mode collapse. |
| Novelty | Novelty Rate | Percentage of valid, unique molecules not present in the training dataset. | Essential for de novo design; must be balanced with similarity to drug-like space. |
| Diversity | Internal Diversity (IntDiv) | Measures structural variety within a generated set using molecular fingerprint similarity (e.g., Tanimoto similarity on ECFP4 fingerprints) [3]. | Higher IntDiv (e.g., 85-89%) indicates broad exploration of chemical space [3]. |
| Scaffold Diversity | Scaffold Hit Rate | Percentage of generated molecules that match a desired target scaffold or scaffold family. | Critical for scaffold-constrained generation tasks. |
| Scaffold Novelty | Percentage of generated molecules containing core scaffolds not present in the training data. | Indicates success in "scaffold hopping" [19]. | |
| Latent Space Quality | Reconstruction Accuracy | Ability of the VAE to encode and then decode a molecule back to its original structure. | Measures the informativeness of the latent representation [20]. |
| Kullback-Leibler (KL) Divergence | Regularization term enforcing the latent space distribution to match a prior (e.g., Gaussian). | Prevents overfitting; balanced with reconstruction loss to avoid posterior collapse [3] [4]. |
The following table summarizes the performance of several contemporary VAE architectures on standard benchmarks, illustrating the trade-offs between different molecular representations and model designs.
Table 2: Performance Benchmark of Representative VAE Architectures
| Model Name | Molecular Representation | Key Architectural Features | Reported Performance Highlights |
|---|---|---|---|
| ScafVAE [43] | Graph | Bond scaffold-based generation, perplexity-inspired fragmentation. | High reconstruction accuracy; successful generation of dual-target drug candidates. |
| NP-VAE [20] | Graph (Junction Tree) | Handles large, complex natural product structures; incorporates chirality. | Higher reconstruction accuracy for large compounds (>500 Da) compared to JT-VAE, HierVAE. |
| PCF-VAE [3] | GenSMILES (SMILES variant) | Mitigates posterior collapse via modified loss function and diversity layer. | Validity: 95-98%; Uniqueness: 100%; Novelty: 94-95%; IntDiv: ~86-89%. |
| STAR-VAE [5] | SELFIES | Transformer-based encoder-decoder; property-guided conditioning. | Matches/exceeds baselines on GuacaMol & MOSES; enables property-aware generation. |
| Conditional β-VAE [50] | SMILES | Disentangled latent space; mutual information training. | State-of-the-art (SOTA) results on penalized LogP (104.29) and QED (0.948) optimization. |
| TGVAE [4] | Graph | Combines Transformer and GNN; addresses over-smoothing & posterior collapse. | Generates larger, more diverse collections of previously unexplored structures. |
Experimental Protocols for Evaluation
This section outlines detailed, step-by-step protocols for key experiments in assessing VAE performance, from standard benchmark evaluations to advanced multi-objective optimization.
Protocol 1: Standardized Benchmarking on GuacaMol/MOSES
Purpose: To objectively compare the chemical space exploration and scaffold diversity of a target VAE model against established baselines under standardized conditions. Principle: The GuacaMol and MOSES benchmarks provide curated datasets and predefined metrics to evaluate fundamental generative model properties [5] [53].
Data Preparation:
- Acquire the training dataset (e.g., ZINC Clean Leads for MOSES, GuacaMol training set) and the benchmark's test set.
- Preprocess molecules: standardize using RDKit, filter by molecular weight (e.g., 250-500 Da) and other drug-likeness criteria, and generate canonical SMILES or SELFIES strings.
Model Training & Generation:
- Train the target VAE model on the benchmark's training dataset. Adhere to recommended hyperparameters for the specific VAE architecture.
- After training, sample 10,000-30,000 latent vectors from the prior distribution ( N(0,I) ) and decode them into molecular structures.
Metric Calculation:
- Validity: Use RDKit to check the chemical validity of each generated string.
- Uniqueness & Novelty: Calculate the percentage of unique molecules and those not present in the training set.
- Internal Diversity: For all valid, unique generated molecules, compute the average pairwise Tanimoto similarity based on ECFP4 fingerprints. Internal Diversity = 1 - average pairwise similarity.
- Scaffold Analysis: Use the Bemis-Murcko method (RDKit) to extract the core scaffold of each generated molecule. Calculate the number of unique scaffolds and the Scaffold Novelty rate.
Results Interpretation: Compare calculated metrics against published baselines (e.g., JT-VAE, GrammarVAE) to determine competitive performance.
Protocol 2: Scaffold-Conditioned Generation and Analysis
Purpose: To explicitly evaluate a model's capability for "scaffold hopping"–generating novel compounds that retain a specific core scaffold while exploring diverse decorations. Principle: Models like ScafVAE and JT-VAE are inherently designed for scaffold-aware generation [43] [20]. This protocol tests this capability.
Scaffold Selection and Latent Space Definition:
- Select a target scaffold of interest (e.g., a benzodiazepine core).
- Identify a set of 50-100 molecules from the training data that contain this scaffold.
- Encode these molecules using the trained VAE to obtain their latent vectors, ( Z_{\text{scaffold}} ).
Conditional Generation:
- Define the scaffold region in latent space, for example, by calculating the centroid ( \mu{\text{scaffold}} ) and covariance matrix of ( Z{\text{scaffold}} ).
- Sample new latent vectors, ( z{\text{new}} ), from a multivariate Gaussian distribution defined by ( \mu{\text{scaffold}} ) and the covariance matrix, or by sampling within the convex hull of ( Z_{\text{scaffold}} ).
- Decode ( z_{\text{new}} ) to generate new molecular structures.
Analysis of Generated Molecules:
- Scaffold Fidelity: Use substructure matching (RDKit) to verify that all generated molecules contain the target scaffold.
- Side-Chain Diversity: For the molecules passing the fidelity check, extract and cluster the side-chain decorations to quantify their structural diversity.
- Property Analysis: Calculate key physicochemical properties (e.g., QED, SAscore, LogP) for the generated molecules and compare them to the original reference set to assess optimization.
Protocol 3: Multi-Objective Latent Space Optimization (LSO)
Purpose: To optimize generated molecules for multiple, potentially conflicting, properties simultaneously (e.g., binding affinity, solubility, low toxicity). Principle: Latent space optimization leverages the continuous nature of the VAE's latent representation to perform gradient-based or Bayesian optimization for desired properties [53].
Initial Model and Predictor Setup:
- Pre-train the VAE on a large, drug-like dataset (e.g., ZINC or PubChem).
- Train surrogate property prediction models (e.g., neural networks) on the latent space to predict each target property of interest (e.g., pLogP, QED, docking score).
Iterative Weighted Retraining:
- Generate & Score: Use the current VAE to generate a large pool of candidate molecules. Score them using the property predictors.
- Pareto Ranking: Rank all molecules (generated candidates + original training data) based on their Pareto efficiency for the multiple objectives. Molecules that are non-dominated receive the highest rank.
- Weight Assignment: Assign a weight to each molecule in the training set based on its Pareto rank. Higher-ranked molecules receive exponentially higher weights.
- Model Retraining: Retrain the VAE on the weighted training dataset. This reshapes the latent space to be more densely populated in high-performing regions.
- Iteration: Repeat steps a-d for a fixed number of iterations or until performance plateaus.
The Scientist's Toolkit: Research Reagent Solutions
This section catalogs the essential computational tools, datasets, and software libraries required to implement the protocols and conduct research in this field.
Table 3: Essential Research Reagents for VAE Molecular Generation Research
| Reagent / Resource | Type | Function and Application | Example / Source |
|---|---|---|---|
| RDKit | Software Library | Open-source cheminformatics toolkit; used for molecule validity checks, descriptor calculation, fingerprint generation, and scaffold analysis. | https://www.rdkit.org |
| ZINC Database | Dataset | A freely available database of commercially available compounds; provides standard training sets for generative models (e.g., ZINC-250k). | https://zinc.docking.org |
| PubChem | Dataset | A large, public repository of chemical substances and their biological activities; used for large-scale pre-training (e.g., ~79M molecules) [5]. | https://pubchem.ncbi.nlm.nih.gov |
| GuacaMol Benchmark | Framework & Dataset | A benchmark suite for de novo molecular design, providing standardized metrics and datasets for evaluating model performance [53]. | https://github.com/BenevolentAI/guacamol |
| MOSES Benchmark | Framework & Dataset | A benchmarking platform (Molecular Sets) to train and evaluate molecular generative models, promoting reproducibility. | https://github.com/molecularsets/moses |
| ECFP4 Fingerprints | Computational Method | Extended-Connectivity Fingerprints; a circular fingerprint used to represent molecular structures for similarity and diversity calculations. | Implemented in RDKit |
| SELFIES | Molecular Representation | A string-based representation that guarantees 100% syntactic validity, overcoming a major limitation of SMILES [5]. | https://github.com/aspuru-guzik-group/selfies |
| PyTor / TensorFlow | Software Library | Deep learning frameworks used for implementing and training VAE architectures. | https://pytorch.org/ https://www.tensorflow.org/ |
Conclusion
Variational Autoencoders have firmly established themselves as a cornerstone technology in generative molecular design, demonstrating significant capabilities in navigating vast chemical spaces for drug discovery. The synthesis of insights from foundational principles to advanced architectures reveals a trajectory toward increasingly sophisticated models that effectively balance structural validity, diversity, and target-specific optimization. Key advancements in graph-based representations, hybrid modeling, and robust benchmarking are systematically addressing long-standing challenges such as posterior collapse and limited novelty. Looking forward, the integration of VAEs with multimodal data, large-scale pretraining, and automated closed-loop design systems promises to accelerate the discovery of novel therapeutic candidates. The convergence of these technologies is poised to reshape preclinical drug development, enabling more efficient exploration of underexplored chemical territories and ultimately contributing to the development of precision medicines. Future research should focus on improving model interpretability, incorporating synthetic accessibility, and enhancing the handling of complex 3D molecular properties to fully realize the potential of AI-driven molecular science in clinical translation.
References
- 1. Variational Autoencoder (VAE): The Ultimate Guide for 2025 [https://www.dhiwise.com/post/variational-autoencoder-guide]
- 2. Variational AutoEncoders [https://www.geeksforgeeks.org/machine-learning/variational-autoencoders/]
- 3. PCF-VAE: posterior collapse free variational autoencoder ... [https://www.nature.com/articles/s41598-025-14285-5]
- 4. Transformer graph variational autoencoder for generative ... [https://www.sciencedirect.com/science/article/pii/S0006349525000359]
- 5. STAR-VAE: Latent Variable Transformers for Scalable and ... [https://arxiv.org/html/2511.02769v1]
- 6. What Is Latent Space? | IBM [https://www.ibm.com/think/topics/latent-space]
- 7. How to Sample From Latent Space With Variational ... [https://hackernoon.com/how-to-sample-from-latent-space-with-variational-autoencoder]
- 8. Ensemble Visualization With Variational Autoencoder [https://arxiv.org/html/2509.13000v1]
- 9. Molecular representation learning: cross-domain foundations ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00170f]
- 10. Evidence, KL-divergence, and ELBO - Massimiliano Patacchiola [https://mpatacchiola.github.io/blog/2021/01/25/intro-variational-inference.html]
- 11. Snap-Fast Molecular Generation with Latent Variational ... [https://arxiv.org/html/2508.05411]
- 12. A scalable variational inference approach for increased ... [https://www.nature.com/articles/s41588-024-02044-7]
- 13. PMF-GRN: a variational inference approach to single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11003171/]
- 14. The Reparameterization in Trick Variational Autoencoders [https://www.baeldung.com/cs/vae-reparameterization]
- 15. “ Reparameterization ” trick in Variational | Medium Autoencoders [https://medium.com/data-science/reparameterization-trick-126062cfd3c3]
- 16. Reparameterization trick [https://en.wikipedia.org/wiki/Reparameterization_trick]
- 17. Targeted molecular generation with latent reinforcement ... [https://www.nature.com/articles/s41598-025-99785-0]
- 18. Reinforcement Learning-Inspired Molecular Generation ... [https://www.sciencedirect.com/science/article/abs/pii/S000925092501396X]
- 19. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 20. Variational autoencoder-based chemical latent space for ... [https://www.nature.com/articles/s42004-023-01054-6]
- 21. Molecular generative model based on conditional variational ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-018-0286-7]
- 22. Simplified Molecular Input Line Entry System [https://en.wikipedia.org/wiki/Simplified_Molecular_Input_Line_Entry_System]
- 23. 3. SMILES - A Simplified Chemical Language [https://www.daylight.com/dayhtml/doc/theory/theory.smiles.html]
- 24. SMILES (Simplified Molecular Input Line Entry System) [https://hunterheidenreich.com/notes/formats/computational-chemistry/smiles/]
- 25. SMILES vs. Graph representation in deep learning [https://chemistry.stackexchange.com/questions/128149/smiles-vs-graph-representation-in-deep-learning]
- 26. Comparison of SMILES and molecular graphs as the ... [https://www.sciencedirect.com/science/article/abs/pii/S0169743911001560]
- 27. Latent Space Characterization of Autoencoder Variants [https://arxiv.org/html/2412.04755v1]
- 28. Conditional Latent Space Molecular Scaffold Optimization for Accelerated Molecular Design [https://arxiv.org/html/2411.01423]
- 29. model Generative on junction tree variational autoencoder for... based [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00681-4]
- 30. Variational autoencoder- based chemical latent space for large... [https://www.nature.com/articles/s42004-023-01054-6?error=cookies_not_supported&code=ac8d2564-e875-44e8-904b-f70c0687cd4e]
- 31. Hybridization of SMILES and chemical-environment-aware ... [https://www.nature.com/articles/s41598-025-01890-7]
- 32. Generating Molecules using a Char-RNN in Pytorch [https://medium.com/@sunitachoudhary103/generating-molecules-using-a-char-rnn-in-pytorch-16885fd9394b]
- 33. Generating Molecules using CharRNN [https://blog.bayeslabs.co/2019/07/04/Generating-Molecules-using-Char-RNN-in-Pytorch.html]
- 34. Grammar Variational Autoencoder (GVAE) [https://www.emergentmind.com/topics/grammar-variational-autoencoder-gvae]
- 35. Learning Molecular Dynamics with Simple Language ... [https://arxiv.org/abs/2004.12360]
- 36. Deep generative model of constructing chemical latent ... [https://chemrxiv.org/engage/chemrxiv/article-details/646881bbfb40f6b3eee2e089]
- 37. Variational autoencoder-based chemical latent space for ... [https://pubmed.ncbi.nlm.nih.gov/37973971/]
- 38. Deep Learning-Driven Molecular Generation and ... [https://www.mdpi.com/2076-3417/15/7/3640]
- 39. Understanding Conditional Variational Autoencoders | Medium [https://medium.com/data-science/understanding-conditional-variational-autoencoders-cd62b4f57bf8]
- 40. ICVAE: Interpretable Conditional Variational Autoencoder ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12071458/]
- 41. Target-aware 3D molecular generation based on guided ... [https://www.nature.com/articles/s41467-025-63245-0]
- 42. GitHub - gozsoy/ conditional - vae : Tensorflow implementation of... [https://github.com/gozsoy/conditional-vae]
- 43. Multi-objective drug design with a scaffold-aware variational ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc08736d]
- 44. Phenotypic Profile-Informed Generation of Drug-Like ... [https://arxiv.org/html/2506.02051v1]
- 45. Inside ELRIG's Drug Discovery 2025 [https://www.drugtargetreview.com/article/190237/inside-elrigs-drug-discovery-2025/]
- 46. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 47. Posterior Collapse as a Phase Transition in Variational ... [https://arxiv.org/html/2510.01621v2]
- 48. Preventing Posterior Collapse with DVAE for Text Modeling [https://pmc.ncbi.nlm.nih.gov/articles/PMC12026048/]
- 49. What is "posterior collapse" phenomenon? [https://datascience.stackexchange.com/questions/48962/what-is-posterior-collapse-phenomenon]
- 50. Conditional $β$-VAE for De Novo Molecular Generation [https://arxiv.org/abs/2205.01592]
- 51. Variational autoencoders - Matthew N. Bernstein [https://mbernste.github.io/posts/vae/]
- 52. Variational Autoencoder Tutorial: VAEs Explained [https://www.codecademy.com/article/variational-autoencoder-tutorial-vaes-explained]
- 53. Multi-objective latent space optimization of generative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11573897/]
- 54. Secrets of the Variational AutoEncoder(VAE ) [https://medium.com/data-science-collective/secrets-of-the-variational-autoencoder-vae-appreciation-without-the-apprehension-9389095d461c]
- 55. Improving VAE based molecular representations for ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-022-00648-x]
- 56. Exploring Over-Smoothing in Graph Neural Networks (GNNs) [https://medium.com/@hellorahulk/exploring-over-smoothing-in-graph-neural-networks-gnns-e311b8eb8a85]
- 57. Over smoothing issue in graph neural network [https://towardsdatascience.com/over-smoothing-issue-in-graph-neural-network-bddc8fbc2472/]
- 58. Mitigating over-smoothing in Graph Neural Networks for ... [https://www.sciencedirect.com/science/article/abs/pii/S0950705125006616]
- 59. Graph Neural Networks (with Proper Weights) Can Escape ... [https://proceedings.mlr.press/v260/zhuo25a.html]
- 60. Unifying over-smoothing and over-squashing in graph ... [https://arxiv.org/abs/2309.02769]
- 61. Over-Smoothing and Over-Squashing in GNNs [https://www.sciopen.com/article/10.26599/BDMA.2023.9020019]
- 62. Preventing Oversmoothing in VAE via Generalized ... [https://arxiv.org/abs/2102.08663]
- 63. GenSMILES: An enhanced validity conscious ... [https://www.sciencedirect.com/science/article/abs/pii/S095070512300179X]
- 64. Towards Unified Latent Space for 3D Molecular ... [https://arxiv.org/html/2503.15567v1]
- 65. Leveraging tree-transformer VAE with fragment ... [https://www.nature.com/articles/s42004-025-01640-w]
- 66. PCF-VAE: posterior collapse free variational autoencoder for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12488873/]
- 67. Ig-VAE: Generative modeling of protein structure by direct ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9269947/]
- 68. Generative Deep Learning for de Novo Drug Design A ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12308806/]
- 69. Generative artificial intelligence based models optimization ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01059-4]
- 70. A generative framework for enhancing drug target ... [https://www.nature.com/articles/s41598-025-01589-9]
- 71. Synthetic Scientific Image Generation with VAE, GAN, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12387873/]
- 72. Molecular Sets (MOSES): A Benchmarking Platform for ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2020.565644/full]
- 73. Molecular Alchemy: How MOSES is Redefining Drug ... [https://pharmafeatures.com/molecular-alchemy-how-moses-is-redefining-drug-discovery-in-the-age-of-ai/]
- 74. A Comprehensive Benchmarking Platform for Deep ... [https://arxiv.org/abs/2505.12848]
- 75. De novo molecular drug design benchmarking - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8372209/]
- 76. Molecular Sets (MOSES): A Benchmarking Platform for ... [https://github.com/molecularsets/moses]
- 77. integration of variational autoencoders and normalizing flows ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12553153/]
- 78. relational graph conditioned variational autoencoder for ... [https://link.springer.com/article/10.1007/s10994-024-06638-4]
- 79. WACV 2025 Open Access Repository [https://openaccess.thecvf.com/content/WACV2025/html/Saha_ARD-VAE_A_Statistical_Formulation_to_Find_the_Relevant_Latent_Dimensions_WACV_2025_paper.html]
- 80. ARD-VAE: A Statistical Formulation to Find the Relevant ... [https://arxiv.org/abs/2501.10901]
- 81. Transformer graph variational autoencoder for generative ... [https://pubmed.ncbi.nlm.nih.gov/39885689/]
- 82. LoG3D: Ultra-High-Resolution 3D Shape Modeling via Local-to-Global... [https://arxiv.org/html/2511.10040v2]
- 83. How to train your VAE [https://arxiv.org/html/2309.13160v3]
- 84. ICVAE: Interpretable Conditional Variational Autoencoder ... [https://www.mdpi.com/1422-0067/26/9/3980]
- 85. Bridging chemical space and biological efficacy: advances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12144013/]