TIDE Assay: A Complete Guide to Tracking Indels in CRISPR Genome Editing
This article provides a comprehensive overview of the Tracking of Indels by Decomposition (TIDE) assay, a widely used method for quantifying genome editing efficiency.
TIDE Assay: A Complete Guide to Tracking Indels in CRISPR Genome Editing
Abstract
This article provides a comprehensive overview of the Tracking of Indels by Decomposition (TIDE) assay, a widely used method for quantifying genome editing efficiency. Tailored for researchers and drug development professionals, it covers the foundational principles of TIDE, its application in quantifying insertions and deletions (indels) from Sanger sequencing data, and step-by-step methodological guidance. The content further explores troubleshooting common issues, optimizing parameters for accurate results, and presents a comparative analysis with other validation techniques like T7E1, ICE, and next-generation sequencing. By synthesizing current research and best practices, this guide serves as an essential resource for validating CRISPR-Cas9, TALEN, and ZFN editing outcomes in biomedical research and therapeutic development.
What is the TIDE Assay? Principles and Applications in Modern Genomics
The advent of CRISPR-Cas9 and other programmable nucleases has revolutionized biological research and therapeutic development, creating an pressing need for accessible methods to quantify their editing efficiency. Tracking of Indels by Decomposition (TIDE) emerged as a breakthrough computational tool that enables rapid, quantitative assessment of genome editing outcomes directly from Sanger sequencing data [1] [2]. Developed by the Bas van Steensel lab, TIDE addresses a critical bottleneck in the genome editing workflow by providing researchers with a simple yet powerful method to decipher the complex mixture of insertion and deletion mutations (indels) generated at a target locus [1] [3].
Unlike next-generation sequencing (NGS), which provides comprehensive data but requires substantial time, cost, and bioinformatics expertise, TIDE leverages the ubiquitous availability and affordability of Sanger sequencing to deliver quantitative indel analysis [4] [2]. The method computationally deconvolves sequencing chromatograms from edited cell populations, reconstructing the spectrum and frequency of different indel mutations without the need for cloning or deep sequencing [1] [5]. This approach has made quantitative editing assessment accessible to laboratories worldwide, accelerating the development and validation of genome editing experiments across diverse biological systems and applications.
Core Principles and Algorithmic Foundation
TIDE operates on a fundamental principle: the sequencing chromatogram from an edited cell population represents a composite signal derived from the weighted contribution of all different indel sequences present in the sample [1]. The algorithm employs a decomposition approach to resolve this complex signal into its constituent parts by comparing the edited sample chromatogram against a reference sequence from an unedited control [1] [2].
The mathematical core of TIDE utilizes non-negative linear regression modeling (NNLS) to identify the combination of indel sequences that best explains the observed sequencing trace data from the edited sample [1] [2]. This method systematically tests potential indel combinations within user-defined parameters, calculating the statistical significance of each detected mutation and providing a goodness-of-fit measure (R² value) to assess the reliability of the decomposition [1]. The algorithm is particularly optimized for detecting small insertions and deletions typically generated by non-homologous end joining (NHEJ) repair of CRISPR-induced double-strand breaks, with a default detection range of up to 10 base pairs, though this parameter can be adjusted by the user [1].
Experimental Workflow and Protocol
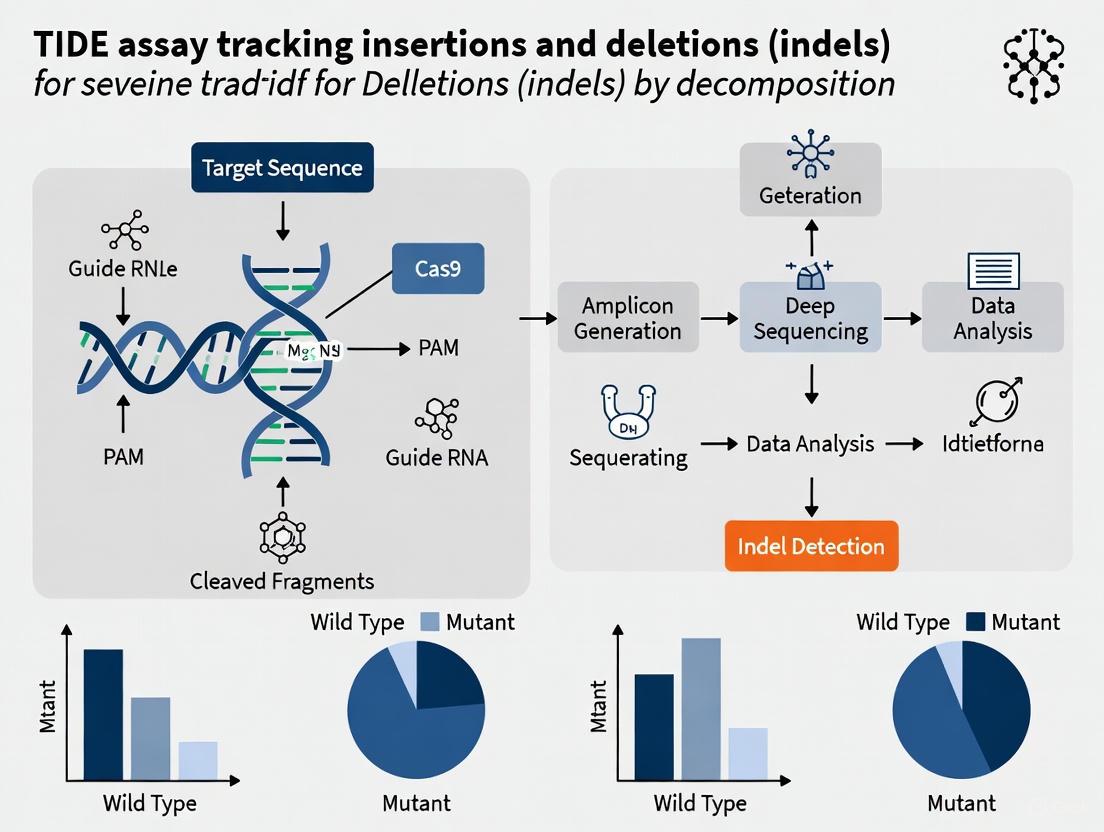
The experimental implementation of TIDE follows a streamlined workflow that can be completed with standard molecular biology reagents and techniques. The process begins with PCR amplification of the target locus from both control (unmodified) and experimentally edited cell populations [1] [3]. The resulting amplicons are then subjected to standard Sanger sequencing using the same primer for both samples. For optimal results, the target region should be sequenced to a length of approximately 700 base pairs, with the Cas9 cut site positioned preferably around 200 base pairs downstream from the sequencing start site [1].
Once sequencing is complete, the raw chromatogram files (in .ab1 or .scf format) are uploaded to the TIDE web tool along with the sgRNA sequence used for editing [1]. The software automatically processes the data through several key steps: First, it aligns the control and test sequences within a defined alignment window to correct for any systematic shifts between the traces. Next, it performs the decomposition analysis within a specified decomposition window downstream of the expected cut site. Finally, it generates a comprehensive report detailing the identified indels, their frequencies, and statistical confidence metrics [1].
Key Parameters and Quality Control Metrics
Successful application of TIDE requires careful attention to several critical parameters and quality metrics. The alignment window, typically set from base 100 to a position 10 base pairs before the expected cut site, ensures proper registration of the test and control sequences [1]. The decomposition window, automatically set to the maximum possible range by default, determines the sequence segment used for indel detection and should encompass the region where sequencing quality remains high [1].
TIDE provides several built-in quality control measures to assess data reliability. The aberrant sequence signal plot visualizes the percentage of non-consensus nucleotide signals at each position, with well-performing samples showing low aberrant signal (<10%) before the break site in both control and test samples, followed by increased signal specifically in the test sample after the break site [1]. The goodness-of-fit (R²) should exceed 0.9 for reliable decomposition, and the statistical significance of each identified indel is calculated using a default p-value threshold of <0.001 [1]. These quality metrics are essential for interpreting TIDE results and ensuring accurate quantification of editing efficiency.
Comparative Performance Analysis of CRISPR Analysis Methods
Head-to-Head Tool Comparison Using Reference Standards
Multiple independent studies have systematically evaluated the performance of TIDE against other CRISPR analysis tools using artificial sequencing templates with predetermined indel frequencies and compositions [2] [6]. These controlled experiments reveal important insights into the relative strengths and limitations of each method across different editing scenarios.
When analyzing simple indel patterns consisting of a few base changes, TIDE and other tools (ICE, DECODR, SeqScreener) demonstrate acceptable accuracy in estimating overall indel frequencies [2]. However, as the complexity of indel patterns increases, significant variability emerges between tools, with each exhibiting different sensitivities to specific types of mutations [2]. A particularly comprehensive comparison using zebrafish gene editing models found that while all four tools accurately estimated net indel sizes, their ability to deconvolute specific indel sequences varied considerably, with DECODR providing the most accurate estimations for the majority of samples [2].
Table 1: Performance Comparison of CRISPR Analysis Methods Using Artificial Reference Standards
| Analysis Tool | Accuracy with Simple Indels | Performance with Complex Indels | Large Deletion Detection | Knock-in Efficiency Analysis |
|---|---|---|---|---|
| TIDE | Acceptable accuracy [2] | Variable performance [2] | Limited (<50 bp) [5] | Requires TIDER extension [3] |
| ICE (Synthego) | High correlation with NGS [4] | Better than TIDE for complex patterns [4] | Capable of detection [4] | Limited capabilities [2] |
| DECODR | Most accurate in study [2] | Superior sequence deconvolution [2] | Improved range [5] | Not specialized [2] |
| NGS | Gold standard [4] [2] | Comprehensive detection [4] | No size limitation [1] | Direct quantification possible [2] |
Performance in Real-World Applications
The performance characteristics observed in controlled studies manifest distinctly in practical research scenarios. A particularly revealing investigation analyzed CRISPR-Cas9 editing in somatic mouse tumor models and discovered high variability in reported indel number, size, and frequency across different software platforms [6]. This study highlighted that software selection significantly impacts experimental conclusions, especially when samples contain larger indels that are common in somatic in vivo editing contexts [6].
When compared to non-sequencing-based methods like the T7 Endonuclease I (T7E1) assay, TIDE demonstrates markedly superior quantitative capabilities [7]. The T7E1 assay tends to have a low dynamic range and frequently misrepresents editing efficiency, particularly for samples with either very low or very high editing rates or those with a single dominant indel [7]. In contrast, TIDE provides more linear quantification across a broader range of editing efficiencies and offers the crucial advantage of identifying the specific sequences of induced mutations [8] [7].
Table 2: Method Comparison Across Practical Research Applications
| Application Context | TIDE Performance | Alternative Methods | Key Considerations |
|---|---|---|---|
| In vivo somatic editing | High variability with large indels [6] | DECODR handles larger indels better [6] [5] | Software choice critical for accurate interpretation [6] |
| High-throughput gRNA screening | Efficient for moderate-scale studies [1] | NGS preferred for large-scale studies [4] | Batch processing available but less streamlined than ICE [1] [4] |
| Therapeutic development | Good for preliminary characterization [8] | ddPCR offers superior quantification [8] | May miss large deletions relevant to safety assessment [1] [5] |
| Academic research | Excellent balance of cost and information [2] | T7E1 cheaper but less quantitative [7] | User-friendly for labs without bioinformatics support [1] [2] |
Limitations and Detection Constraints
Despite its utility, TIDE has several important limitations that researchers must consider when interpreting results. The most significant constraint is its limited ability to detect large deletions exceeding 50 base pairs, a recognized shortcoming that has prompted the development of specialized tools like PtWAVE that extend detection to 200 bp [5]. This limitation is particularly relevant for therapeutic applications where large deletions may have functional consequences [1].
Additionally, TIDE does not naturally capture megabase-scale deletions that can occasionally originate from CRISPR-Cas9-induced double-strand breaks [1]. The method also assumes that the double-strand break occurs at a specific position (between nucleotides 17 and 18 of the sgRNA sequence), which may not account for variations in cutting position across different nuclease platforms [1]. For templated CRISPR-Cas9 experiments involving homology-directed repair, researchers must use TIDER, a modified version of TIDE that requires an additional sequencing trace [3].
Essential Research Reagents and Experimental Solutions
Successful implementation of TIDE analysis requires careful attention to experimental design and reagent selection. The following table outlines key solutions and methodological considerations for optimal TIDE analysis.
Table 3: Essential Research Reagents and Methodological Solutions for TIDE Analysis
| Reagent/Component | Function in TIDE Workflow | Technical Specifications | Quality Considerations |
|---|---|---|---|
| PCR Enzymes | Amplification of target locus from genomic DNA | High-fidelity polymerase recommended | Minimize PCR artifacts and amplification bias [2] |
| Sanger Sequencing | Generation of chromatogram data | ~700 bp amplicon length ideal | Target Phred score >30 in alignment region [1] [5] |
| Control DNA Sample | Reference for decomposition analysis | Unedited sample, same genetic background | Critical for accurate alignment and background subtraction [1] |
| sgRNA Sequence | Input for cut site determination | 20nt guide sequence without PAM | Valid DNA characters only; invalid characters automatically removed [1] |
| Chromatogram Files | Raw data for analysis | .ab1 or .scf format supported | Ensure good sequence quality with low aberrant signal before break site [1] |
Method Selection Framework for Genome Editing Analysis
The optimal choice of genome editing analysis method depends on multiple factors, including experimental goals, resource constraints, and required detection capabilities. The following decision framework illustrates the method selection process based on common research scenarios.
Emerging Alternatives and Future Directions
The landscape of CRISPR analysis tools continues to evolve, with new algorithms addressing specific limitations of established methods like TIDE. The recently developed PtWAVE (Progressive-type Wide-range Analysis of Varied Edits) software significantly expands detection capabilities for large indels, enabling reliable identification of deletions up to 200 bp while maintaining analytical precision [5]. This extension is particularly valuable for applications where large deletions are biologically or therapeutically relevant.
Another emerging trend is the integration of multiple analysis modalities to leverage their complementary strengths. For example, combining the cost-effectiveness of TIDE with the precise quantification of droplet digital PCR (ddPCR) provides a balanced approach for therapeutic development [8]. Meanwhile, the ongoing refinement of TIDER for templated editing analysis demonstrates how core decomposition algorithms can be adapted to specialized applications [3].
As CRISPR technology advances toward clinical applications, analysis methods must evolve correspondingly. Future developments will likely focus on improving detection of complex editing outcomes, including translocations and structural variations, while maintaining the accessibility that has made tools like TIDE invaluable to the research community. The continued benchmarking of these tools against gold-standard NGS approaches remains essential for establishing their appropriate use across diverse genome editing contexts [2] [6] [7].
The advent of programmable nucleases, such as CRISPR-Cas9, has revolutionized biological research and therapeutic development by enabling precise genome modifications. These editing tools primarily induce double-strand breaks at specific genomic loci, which are subsequently repaired by cellular mechanisms like non-homologous end joining (NHEJ). This repair process often results in a spectrum of small insertions and deletions (indels) at the target site. Characterizing these indels is crucial for assessing editing efficiency and specificity, yet the heterogeneous mixture of editing outcomes in a cell population presents a significant analytical challenge. Sanger sequencing, a longstanding workhorse of molecular biology, generates chromatogram traces that become complex and uninterpretable by eye when derived from such heterogeneous samples. Computational deconvolution approaches transform these complex chromatograms into quantitative insights about editing outcomes, bridging the gap between simple enzymatic assays and expensive next-generation sequencing.
The Fundamental Principle of Trace Decomposition
At its core, chromatogram deconvolution is a computational strategy designed to resolve complex Sanger sequencing data from a mixed sample into its constituent sequences. A standard Sanger sequencing reaction from a clonal source produces a clean chromatogram with single peaks at each position. In contrast, a PCR-amplified sample from a genome-edited cell pool contains a mixture of DNA fragments with different indels, resulting in a chromatogram with overlapping signals and multiple peaks after the cut site. The deconvolution algorithm tackles this by comparing the edited sample's trace data to a control (unmodified) trace.
The process typically involves several key steps. First, the algorithm aligns the control and test sequences, identifying the nuclease cut site—usually located 3-4 bases upstream of the protospacer adjacent motif (PAM) for Cas9. Next, within a defined "decomposition window" downstream of the cut site, the algorithm tests a comprehensive set of possible indel combinations (within a user-defined size range, typically up to 10-15 bp) to find the optimal linear combination that reconstructs the observed trace data from the edited sample. This is achieved through non-negative linear regression, which estimates the frequency of each indel event that, when summed together, best explains the complex trace pattern. The output is a quantitative profile of the predominant indels and their respective frequencies in the population.
Comparative Analysis of Deconvolution Tools
Several computational tools have been developed that implement variations of the core deconvolution principle, each with unique features and performance characteristics. The following table provides a structured comparison of the major tools available to researchers.
Table 1: Key Software Tools for Deconvolving Sanger Sequencing Chromatograms
| Tool Name | Primary Function | Input Requirements | Key Outputs | Notable Features |
|---|---|---|---|---|
| TIDE (Tracking of Indels by Decomposition) [1] [9] [3] | Quantifies indel spectrum from non-templated editing. | - sgRNA sequence- Control sample trace file- Edited sample trace file | - Indel frequencies and spectrum- Goodness of fit (R²)- Statistical significance (p-value) | - Interactive web tool- Established, widely cited method- Specific module for +1 insertions |
| TIDER (Tracking of Insertions, Deletions, and Recombination) [3] | Quantifies both templated (HDR) and non-templated (NHEJ) edits. | - sgRNA sequence- Control sample trace file- Edited sample trace file- Reference (donor) trace file | - Frequency of templated mutation- Spectrum of non-templated indels | - Extension of TIDE for HDR analysis- Requires one additional sequencing trace |
| ICE (Inference of CRISPR Edits) [4] [10] | Analyzes CRISPR data to determine indel abundance and types. | - Sanger sequencing data (.ab1) from control and edited samples | - ICE Score (indel frequency)- Knockout Score (frameshift focus)- Detailed indel distribution | - User-friendly interface- Batch upload capability- High correlation with NGS data (R² = 0.96) |
| Tracy [11] | Comprehensive tool for basecalling, alignment, assembly, and deconvolution. | - Chromatogram files (.ab1, .scf) | - Variant calls in BCF format- Allelic fractions- Decomposition error estimates | - Standalone command-line application & web apps- Generic indel detection (not guide-specific) |
| SuperDecode [12] | Integrated toolkit for analyzing mutations from multiple sequencing strategies. | - Sanger, short-read, or long-read sequencing data from PCR amplicons | - Decoded editing outcomes- Versatile for different edits and ploidies | - Multi-module platform (DSDecodeMS, HiDecode, LaDecode)- Analyzes base and prime editing outcomes |
| DECODR (Deconvolution of Complex DNA Repair) [10] | Deconvolutes complex DNA repair outcomes from Sanger traces. | - Sanger sequencing data | - Indel frequencies- Net indel sizes- Identified indel sequences | - Reported high accuracy in systematic comparisons [10] |
Performance and Experimental Validation
Independent studies have systematically compared the accuracy of these tools using artificial sequencing templates with predetermined indels. The performance can vary based on the complexity of the editing outcomes.
Table 2: Performance Comparison Based on Experimental Data [10]
| Tool | Performance with Simple Indels | Performance with Complex Indels/Knock-ins | Notable Strengths |
|---|---|---|---|
| TIDE | Accurate frequency estimation for few base changes. | Variable performance; more divergent estimates. | Reliable for basic NHEJ analysis; TIDER module for knock-ins. |
| ICE | High correlation with NGS validation. | Capable of detecting larger indels. | User-friendly; provides a dedicated Knockout Score. |
| DECODR | Highly accurate estimation for most samples. | Most accurate for identifying complex indel sequences. | Top performer for precise sequence identification [10]. |
| SeqScreener | Acceptable accuracy for simple cases. | Performance varies with complexity. | Integrated into a commercial platform (Thermo Fisher). |
A critical finding is that while all tools can estimate the net indel size with good accuracy, their ability to correctly deconvolute the exact indel sequences varies significantly. DECODR has been identified as the most accurate for this purpose in several comparisons [10]. For knock-in analysis, TIDER, a variant of TIDE, is specifically designed and often outperforms other general tools for quantifying templated mutations [10].
Detailed Experimental Workflow: From Cells to Analysis
The standard workflow for using tools like TIDE involves a series of wet-lab and computational steps, each critical for obtaining reliable results.
Sample Preparation and Sequencing
- Cell Transfection and Editing: Transfert your target cells with the CRISPR-Cas9 components (e.g., Cas9 and sgRNA expression vectors). Include a control sample that is not edited (e.g., transfected without Cas9 or without the sgRNA).
- Genomic DNA Extraction: Harvest cells, typically 3 days post-transfection, and isolate genomic DNA using a standard kit [9].
- PCR Amplification: Design primers that flank the targeted editing site and amplify the region from both the control and edited genomic DNA samples. The amplicon should be large enough to provide sufficient sequence context; a ~700 bp fragment with the cut site located ~200 bp downstream from the sequencing primer is recommended [1].
- Sanger Sequencing: Purify the PCR products and perform standard capillary sequencing using one of the PCR primers. The resulting chromatogram files (e.g., in .ab1 or .scf format) for the control and test samples are the primary inputs for deconvolution analysis.
Computational Analysis with TIDE
The following diagram illustrates the core computational process of the TIDE algorithm.
- Alignment and Cut Site Identification: The TIDE software first aligns the provided sgRNA sequence to the control sequence to determine the precise location of the expected Cas9 cut site (between nucleotides 17 and 18 of the guide sequence) [1]. It then aligns the control and test sample sequences in a window upstream of the cut site to correct for any sequencing offsets [9].
- Trace Decomposition: The algorithm defines a "decomposition window" downstream of the cut site. Within this window, it tests a set of possible indel mutations (default size up to 10 bp). The decomposition process uses the quantitative peak height data from the sequencing trace to find the linear combination of these indels that best reconstructs the trace data from the edited sample [1] [9].
- Output and Quality Control: The tool generates an indel spectrum plot showing the identified mutations and their frequencies. It also provides an R² value as a measure of goodness-of-fit. A key quality control step involves examining the "Aberrant sequence signal" plot, which visualizes the background noise and the increase in signal divergence after the cut site in the edited sample [1]. An average aberrant signal below 10% before the break site and an R² > 0.9 are indicators of a reliable decomposition [1].
Essential Research Reagent Solutions
The following table catalogs the key reagents and materials required to successfully execute a TIDE analysis from start to finish.
Table 3: Essential Research Reagents and Materials for TIDE Analysis
| Item | Function/Description | Example Product/Catalog |
|---|---|---|
| Programmable Nuclease | Induces the targeted double-strand break. | e.g., CRISPR-Cas9 system (PX330 vector, Addgene #42230) [9]. |
| Cell Culture Reagents | For growing and maintaining cells pre- and post-transfection. | Culture medium, fetal bovine serum (FBS), antibiotics [9]. |
| Transfection Reagent | Delivers editing constructs into cells. | Lipofectamine 2000 for some cell lines; Nucleofector for hard-to-transfect cells [9]. |
| Genomic DNA Isolation Kit | Purifies high-quality DNA for PCR amplification. | e.g., ISOLATE II Genomic DNA Kit (Bioline) [9]. |
| PCR Enzymes & Master Mix | Amplifies the target locus from genomic DNA. | e.g., MyTaq Red Mix (Bioline) [9]. |
| PCR Purification Kit | Cleans up PCR products before sequencing. | e.g., ISOLATE II PCR and Gel Kit (Bioline) [9]. |
| Sanger Sequencing Service/Kit | Generates the raw chromatogram data for analysis. | e.g., BigDye Terminator v3.1 (Applied Biosystems) [9]. |
| TIDE Web Tool | The software that performs the trace deconvolution. | https://tide.nki.nl/ [1] [3]. |
Deconvolution of Sanger sequencing chromatograms represents a powerful and cost-effective principle that has democratized the analysis of genome editing experiments. Tools like TIDE, ICE, and DECODR leverage this principle to provide researchers with quantitative data on editing efficiency and mutation spectra from routine laboratory data. While TIDE established the foundational method, newer tools have emerged, with DECODR showing superior accuracy for resolving complex indel sequences and TIDER specializing in knock-in analysis [10]. The choice of tool should therefore be guided by the specific editing context and the required level of detail.
Looking forward, the integration of artificial intelligence and deep learning into bioinformatics is poised to further enhance the accuracy and capabilities of these analytical methods. As genome editing becomes increasingly central to therapeutic drug development, robust, accessible, and reliable validation methods will remain the cornerstone of scientific rigor and reproducibility.
Tracking of Indels by Decomposition (TIDE) is a computational method designed for the rapid quantitative assessment of mutations generated by genome editing tools. First described by Brinkman and colleagues, TIDE provides a simple and accurate assay to determine the spectrum and frequency of targeted indels (insertions and deletions) in a pool of cells edited by CRISPR/Cas9, TALENs, or ZFNs [3] [13]. The method revolutionized the field by enabling researchers to move beyond qualitative or semi-quantitative assays to a more precise, sequence-based quantification without requiring next-generation sequencing. TIDE operates by analyzing standard Sanger sequencing traces through a decomposition algorithm that reconstructs the mixed sequence signals resulting from heterogeneous indel patterns [3] [1]. This approach has become particularly valuable for preliminary screening and optimization of genome editing conditions, offering an optimal balance between cost, time, and informational output for many experimental scenarios.
The core principle of TIDE involves comparing sequence chromatograms from edited samples against a wild-type control. The algorithm identifies discrepancies in the sequencing traces downstream of the nuclease cut site, deconvoluting the complex mixture of sequences into quantifiable indel frequencies and types [1]. A key advantage of this method is its minimal requirements—only standard molecular biology reagents are needed, involving just one pair of PCR reactions and one pair of standard capillary ("Sanger") sequencing reactions [3]. The accessibility of TIDE through a user-friendly web tool has further contributed to its widespread adoption across the genome editing community.
TIDE Methodology and Workflow
Core Protocol and Technical Requirements
The TIDE protocol consists of three straightforward steps, making it accessible to laboratories with standard molecular biology capabilities. First, researchers perform PCR amplification of the target region from both control and edited genomic DNA. It is recommended to sequence a DNA stretch of approximately 500-700 base pairs enclosing the designed editing site, with the projected break site positioned preferably about 200 base pairs downstream from the sequencing start site [1] [13]. This positioning ensures sufficient high-quality sequence data for robust analysis both upstream and downstream of the cut site.
Second, the PCR products are subjected to standard Sanger sequencing. The resulting sequence trace files (in .ab1 or .scf format) are then analyzed using the TIDE web tool [13]. The algorithm requires the user to input the guide RNA sequence used for editing (for CRISPR systems) or information about the target site (for TALENs and ZFNs). TIDE assumes that for CRISPR/Cas9, the double-strand break is induced between nucleotides 17 and 18 in the sgRNA sequence, counting upstream from the PAM site [1].
The TIDE algorithm employs several key parameters that can be adjusted by users. The alignment window determines the sequence segment used to align control and test samples, typically set with a left boundary of 100 base pairs to avoid poor-quality sequence at the read start. The decomposition window defines the segment used for indel analysis, positioned downstream of the break site. The indel size range sets the maximum size of insertions and deletions modeled in the decomposition, with a default value of 10 base pairs. Finally, the p-value threshold provides a significance cutoff for decomposition, typically set at p < 0.001 [1].
Key Research Reagent Solutions
The successful implementation of TIDE requires several essential research reagents and materials. The table below details these key components and their functions in the TIDE workflow.
| Research Reagent/Material | Function in TIDE Workflow | Specifications & Notes |
|---|---|---|
| Genomic DNA | Template for PCR amplification of target region | Minimum 1000 cells recommended for comprehensive mutation sampling; standard isolation kits sufficient [13] |
| PCR Primers | Amplification of region surrounding nuclease cut site | Should flank ~500-700 bp region with cut site ~200 bp from sequencing start; standard desalted purity sufficient [1] [13] |
| PCR Master Mix | Amplification of target region | Standard mixes containing buffer, Taq polymerase, dNTPs; high-fidelity polymerases recommended [13] |
| Sanger Sequencing Reagents | Generation of sequence traces | BigDye terminator chemistry or commercial sequencing services; .ab1 or .scf file format required [13] |
| TIDE Web Tool | Analysis of sequencing traces | Freely accessible at https://tide.nki.nl; requires guide RNA sequence and sequencing files [3] [1] |
Performance Comparison of TIDE with Alternative Methods
Direct Comparison with Other Computational Tools
Multiple studies have systematically compared TIDE with other computational tools for analyzing genome editing outcomes. A 2024 systematic comparison evaluated TIDE alongside ICE (Inference of CRISPR Edits), DECODR, and SeqScreener using artificial sequencing templates with predetermined indels [2]. The results demonstrated that these tools could estimate indel frequency with acceptable accuracy when the indels were simple and contained only a few base changes. However, the estimated values became more variable among tools when sequencing templates contained more complex indels or knock-in sequences [2].
Another comparative analysis in 2023 highlighted substantial variability in the number, size, and frequency of indels reported by different software platforms, including TIDE, Synthego's ICE, DECODR, and Indigo, when analyzing the same samples from somatic CRISPR/Cas9 tumor models [6]. This study found that while TIDE performed well for simpler editing outcomes, its accuracy diminished with more complex indel patterns, particularly those containing larger indels which are common in in vivo editing contexts [6].
The performance characteristics across different analysis contexts are summarized in the table below.
| Analysis Context | TIDE Performance | Comparative Performance of Alternatives | Key Study Findings |
|---|---|---|---|
| Simple Indels (few base changes) | Acceptable accuracy for frequency estimation [2] | Similar performance across ICE, DECODR, SeqScreener [2] | All tools effectively estimated net indel sizes with minor variations [2] |
| Complex Indels/Knock-ins | Increased variability in estimation [2] | DECODR provided most accurate estimations for majority of samples [2] | Tools showed variable capability to deconvolute complex indel sequences [2] |
| In Vivo Tumor Models | High variability in reported number, size, frequency of indels [6] | DECODR and ICE showed divergent results from same samples [6] | Platforms reported widely divergent data, especially with larger indels [6] |
| Template-Directed Editing | Not suitable for this application; TIDER should be used instead [3] | TIDER (TIDE variant) outperformed other tools for knock-in efficiency quantification [2] | TIDER specifically designed for templated mutations with donor template [3] |
Comparison with Non-Computational Methods
When compared to non-sequencing based methods like the T7 Endonuclease I (T7E1) assay, TIDE provides significant advantages in quantitative capability and informational output. While T7E1 assays offer a quick and inexpensive way to detect editing events, they are not quantitative and provide no information about the specific sequences of the generated indels [4] [8]. TIDE, in contrast, offers both quantitative frequency data and detailed information about the spectrum of indel sequences present in the edited cell population [4].
A 2025 comparative study of methods for assessing on-target gene editing efficiency highlighted that TIDE and ICE offer more quantitative analysis of gene editing outcomes compared to T7E1 assays, though their accuracy heavily relies on the quality of PCR amplification and sequencing [8]. This study also noted that while digital PCR (ddPCR) provides highly precise quantitative measurements, it requires specialized equipment and probe design, making it less accessible for routine screening applications [8].
Ideal Use Cases and Applications
Optimal Scenarios for TIDE Implementation
Based on comparative performance data, TIDE is particularly well-suited for several specific scenarios in genome editing analysis:
Initial gRNA Screening and Optimization: TIDE provides an ideal solution for rapidly testing multiple guide RNAs during the optimization phase of CRISPR experiments. Its balance of cost-effectiveness and quantitative output makes it practical for screening numerous candidates without the expense of next-generation sequencing [4] [13]. The method efficiently identifies the most effective gRNAs for subsequent experimental use.
Simple Indel Characterization in Homogeneous Cell Populations: When editing results primarily in small, simple indels (a few base pairs) and the edited cell population is relatively homogeneous, TIDE delivers accurate frequency estimates and reliable indel spectrum data [2]. This makes it suitable for many in vitro editing applications where complex mutation patterns are less common.
Routine Assessment of Non-Templated Editing: For experiments focusing exclusively on non-homologous end joining (NHEJ) repair without donor templates, TIDE's streamlined two-sequence workflow offers sufficient information with minimal processing time [3]. Its simplicity compared to TIDER makes it the preferred choice when only indel frequency assessment is required.
Resource-Limited Settings: Laboratories with budget constraints or limited access to next-generation sequencing facilities can utilize TIDE to obtain quantitative editing data using existing Sanger sequencing capabilities [4] [14]. The method brings sophisticated analysis within reach of smaller research groups.
TIDER: Specialized Application for Templated Editing
For editing approaches involving donor templates and homology-directed repair (HDR), the TIDER method provides specialized functionality beyond standard TIDE analysis. TIDER (Tracking of Insertions, DEletions, and Recombination events) represents a modified version of TIDE that estimates the frequency of targeted small nucleotide changes introduced by CRISPR in combination with a donor template, while simultaneously quantifying non-templated indels [3].
The key distinction is that TIDER requires three sequencing traces instead of two—the additional "reference" trace represents the intended edited sequence containing the designer mutation [3]. A 2024 study confirmed that TIDER outperformed other computational tools for estimating knock-in efficiency of short epitope tag sequences [2]. The experimental workflow for TIDER involves a modified PCR strategy to generate the reference sequence, followed by analysis through the dedicated TIDER web tool [13].
Limitations and Alternative Selection Guidelines
Recognized Constraints of TIDE Analysis
Despite its utility in many scenarios, TIDE has several important limitations that researchers must consider when selecting an analysis method:
Limited Detection of Complex Edits: TIDE struggles with accurately quantifying complex indel patterns, particularly those involving larger insertions or deletions. Studies have shown that as indel complexity increases, TIDE's estimation variability grows significantly compared to more advanced tools [2] [6].
Poor Performance with Heterogeneous Samples: In samples with high heterogeneity, particularly those derived from in vivo editing contexts like tumor models, TIDE demonstrates substantial variability in reporting indel frequencies and sizes [6]. The algorithm becomes less reliable when numerous different indels are present at varying frequencies.
Inability to Detect Large Structural Variations: TIDE cannot capture megabase-long deletions or large structural rearrangements that can originate from CRISPR/Cas9-induced double-strand breaks [1]. These significant editing outcomes require alternative detection methods.
No Capability for Template-Directed Analysis: Standard TIDE cannot detect 'designer' mutations generated by homologous recombination using a donor template [3]. For these applications, TIDER must be used instead, requiring additional experimental steps.
Strategic Selection Guide for Genome Editing Analysis Methods
The decision framework below summarizes when to select TIDE versus alternative methods based on specific experimental needs and constraints:
| Experimental Context | Recommended Method | Rationale | Evidence |
|---|---|---|---|
| Routine NHEJ editing with simple indels | TIDE | Optimal balance of speed, cost and information for simple edits | Demonstrates acceptable accuracy for indels with few base changes [2] |
| Editing with donor templates | TIDER | Specifically designed to quantify templated mutations alongside indels | Outperformed other tools for knock-in efficiency quantification [2] |
| Complex editing patterns or in vivo samples | DECODR or NGS | Better handling of complex indel patterns in heterogeneous samples | Provided most accurate estimations for majority of complex samples [2] [6] |
| Maximum accuracy and comprehensive variant detection | Next-Generation Sequencing | Gold standard for sensitivity and comprehensive variant detection | Provides complete spectrum of editing outcomes without decomposition assumptions [4] |
| Rapid, low-cost presence/absence testing | T7E1 Assay | Fastest and cheapest method when quantitative data is not essential | Provides quick results without sequencing but lacks quantitative precision [4] [8] |
TIDE remains a valuable tool in the genome editing analysis toolkit, particularly for initial screening phases and experiments focusing on simple indel patterns resulting from non-templated editing. Its ease of use, cost-effectiveness, and rapid turnaround time make it ideally suited for optimizing guide RNAs and assessing editing efficiency in straightforward experimental contexts. However, researchers working with complex editing outcomes, heterogeneous samples, or templated edits should consider alternative methods such as DECODR, TIDER, or next-generation sequencing to ensure accurate quantification. As the field of genome editing continues to advance with increasingly sophisticated applications, the strategic selection of appropriate analysis methods becomes ever more critical for generating reliable, reproducible results.
The Tracking of Indels by Decomposition (TIDE) assay represents a fundamental methodological advance in the quantitative analysis of genome editing outcomes. Developed to meet the growing need for rapid, cost-effective assessment of CRISPR-Cas9 and other nuclease editing experiments, TIDE provides researchers with a computational approach to deconvolve complex sequencing data from heterogeneous cell populations [1]. Unlike traditional cloning and sequencing methods that require extensive laboratory work, TIDE leverages standard Sanger sequencing traces to quantify the spectrum and frequency of insertions and deletions (indels) introduced at targeted genomic loci [13]. This capability has made TIDE an indispensable tool for researchers across various fields, from basic molecular biology to therapeutic drug development, where precise measurement of editing efficiency is critical for experimental success and reproducibility.
The core innovation of TIDE lies in its algorithmic decomposition of composite sequencing chromatograms from edited cell pools, allowing researchers to move beyond simple efficiency measurements to detailed characterization of editing profiles [1]. By mathematically resolving the mixture of sequences resulting from non-homologous end joining repair of nuclease-induced double-strand breaks, TIDE provides insights that were previously only accessible through more labor-intensive and expensive next-generation sequencing approaches. This balance of detail, accessibility, and cost-effectiveness has cemented TIDE's position in the genome editing workflow, particularly during preliminary testing of guide RNA efficiency and optimization of editing conditions [4].
Core Methodology of TIDE Analysis
Fundamental Workflow and Technical Implementation
The TIDE methodology operates through a carefully designed computational pipeline that transforms raw sequencing data into quantitative indel profiles. The process begins with the alignment of sequencing traces from both control (unedited) and experimental (edited) samples, typically in the ABIF (.ab1) or SCF (.scf) file formats [1]. A critical input parameter is the 20-nucleotide guide RNA sequence immediately upstream of the PAM sequence, which allows TIDE to pinpoint the expected Cas9 cleavage site between nucleotides 17 and 18 of the guide [1]. The algorithm then defines two key analysis windows: an alignment window where control and test sequences are aligned to determine any offset between reads, and a decomposition window where the actual deconvolution of indel sequences occurs [1].
The mathematical core of TIDE employs non-negative linear modeling (NNLS) to decompose the mixed sequencing trace from the edited sample into its constituent sequences [1] [5]. This approach quantifies the relative abundance of each indel variant by comparing the experimental trace to a reference trace generated from the control sample. The model systematically tests possible indel combinations within user-defined parameters, typically focusing on indels up to 10 base pairs in size by default, though this range can be adjusted [1]. Statistical significance is assessed for each detected indel, with a default p-value threshold of < 0.001, and overall model quality is evaluated using an R² value representing the goodness of fit between the computed mixture and the actual sequencing trace [1].
Key Input Requirements and Experimental Design
Successful TIDE analysis depends on appropriate experimental design and quality input materials. The essential inputs include:
Control and experimental sample sequencing traces: Two standard capillary sequencing reactions are required—one from an unedited control sample (e.g., wild-type or mock-transfected cells) and one from the edited cell pool [1] [13]. The control sample provides the reference sequence for comparison.
sgRNA target sequence: A 20nt DNA string (5'-3') representing the sgRNA guide sequence immediately upstream of the PAM sequence, without the PAM itself [1]. This enables precise localization of the expected cleavage site.
High-quality PCR amplicons: Researchers should amplify a DNA stretch of approximately 500-700bp enclosing the designed editing site, with the projected break site preferably located about 200bp downstream from the sequencing start site [1] [13]. This positioning ensures sufficient sequence quality for both alignment and decomposition.
Quality parameters: Optimal sequence quality is crucial, with recommended average aberrant sequence signal strength before the breaksite of <10% for both control and test samples, and a decomposition R² > 0.9 [1]. Sequencing of the opposite strand is recommended to confirm results.
The following workflow diagram illustrates the complete TIDE analysis process from experimental preparation to result interpretation:
Comparative Analysis of TIDE and Alternative Tools
Performance Benchmarking in Controlled Studies
Systematic comparisons of computational tools for analyzing Sanger sequencing-based genome editing outcomes have revealed distinct performance characteristics across platforms. A 2024 study by Lee et al. quantitatively evaluated TIDE alongside ICE, DECODR, and SeqScreener using artificial sequencing templates with predetermined indels [2]. The research demonstrated that these tools could estimate indel frequency with acceptable accuracy when the indels were simple and contained only a few base changes. However, the estimated values became more variable among tools when sequencing templates contained more complex indels or knock-in sequences [2]. Notably, DECODR provided the most accurate estimations of indel frequencies for the majority of samples, consistent with findings from other recent reports [2].
Another comprehensive comparison published in 2025 evaluated TIDE, ICE, ddPCR, and live-cell reporter assays, highlighting the unique strengths and limitations of each method [8]. This study emphasized that while TIDE provides a good balance of accessibility and quantitative capability, its accuracy heavily relies on the quality of PCR amplification and sequencing, which can be a limiting factor under variable experimental conditions or challenging target regions [8]. The research also noted that although all major tools accurately estimated net indel sizes, their capability to deconvolute specific indel sequences exhibited variability with certain limitations [2].
Technical Capabilities and Detection Limits
The comparative performance of TIDE and alternative tools can be understood through their technical specifications and detection capabilities:
Table 1: Comparative Analysis of Sanger Sequencing-Based Indel Detection Tools
| Tool | Primary Algorithm | Max Indel Detection | Key Strengths | Main Limitations |
|---|---|---|---|---|
| TIDE | Non-negative linear modeling (NNLS) [1] [5] | ~10bp (default), adjustable [1] | Simple interface, rapid analysis, statistical significance assessment [1] [4] | Limited for large indels (>50bp), decreased accuracy with complex edits [2] [5] |
| ICE | Proprietary decomposition | Not specified | User-friendly, batch processing, KO score for frameshifts, comparable to NGS (R²=0.96) [4] | Commercial product, less algorithm transparency [4] |
| DECODR | Enhanced TIDE algorithm | Improved for larger indels [5] | Better accuracy for complex indels, handles larger deletion range [2] [5] | Less established in community, potentially more complex parameters [2] |
| PtWAVE | Progressive BIC-optimized NNLS/LASSO [5] | Up to 200bp [5] | Superior large deletion detection, model selection minimizes uncertainty [5] | Newer tool (2025), less extensively validated [5] |
A critical technical limitation observed across TIDE analysis tools is the challenge of detecting large indels. Conventional TIDE and ICE share a common limitation in detecting larger deletions (>50bp) due to their shorter predefined range of possible mutations [5]. This size limitation stems from fundamental constraints in the decomposition algorithms, where extending the detection range introduces greater uncertainty and susceptibility to signal noise [5]. Recent developments like PtWAVE address this limitation through progressive adjustment of mutation sequence patterns and evaluation using Bayesian information criterion, enabling detection of indels up to 200bp while maintaining analysis stability [5].
Experimental Protocols for TIDE Analysis
Sample Preparation and Sequencing Requirements
Robust TIDE analysis begins with careful experimental preparation and quality control. The recommended protocol involves:
Guide RNA Design and Transfection: Design sgRNAs using established online tools (e.g., CRISPR MIT, ChopChop, or DeskGen) [13]. Transfert cells with CRISPR components appropriate for your experimental system, using appropriate controls (e.g., transfected without Cas9 or without sgRNA) [1].
Genomic DNA Isolation: Isolate genomic DNA 1-3 days post-transfection using standard kits or phenol/chloroform extraction [13]. A minimum of 1000 cells should be processed to ensure comprehensive sampling of the mutation complexity.
PCR Amplification: Design primers flanking the target site to amplify a 500-700bp fragment enclosing the editing site [1] [13]. The projected break site should be located approximately 200bp downstream from the sequencing start site to ensure optimal sequence quality around the critical region. Use high-fidelity polymerases and optimize cycle numbers to minimize PCR artifacts.
Sanger Sequencing: Purify PCR products and prepare for Sanger sequencing using the same primers as for amplification [13]. Sequence control and experimental samples in parallel to minimize technical variation. Save sequence trace files in .ab1 or .scf format for TIDE analysis.
For template-directed editing experiments, TIDER requires an additional reference DNA sample prepared through a two-step PCR amplification that incorporates the expected HDR outcome, enabling quantification of precise edits against the background of non-templated indels [13].
TIDE Analysis Parameters and Quality Assessment
Once sequencing is complete, TIDE analysis proceeds through these methodical steps:
Data Upload: Submit the control and test sample sequencing files (.ab1 or .scf) to the TIDE web tool (https://tide.nki.nl) [1] [3]. Enter the 20nt sgRNA sequence (without PAM) to define the expected cleavage site.
Parameter Optimization:
- Alignment Window: Typically set with left boundary at 100bp (to avoid poor-quality sequence start) and right boundary automatically set at break site - 10bp [1].
- Decomposition Window: Default uses the largest possible window for robust estimation; manually adjust if sequence quality is locally poor.
- Indel Size Range: Default is 10bp; increase if larger indels are expected.
- P-value Threshold: Default < 0.001; adjust based on required stringency.
Quality Control Assessment: Evaluate the Aberrant Sequence Signal plot to verify [1]:
- Low, equally distributed aberrant signal in control sample (black line)
- Low signal before breaksite with increased signal downstream in test sample (green line)
- Signal increase around expected cut site (blue dotted line)
- R² > 0.9 for decomposition result indicates good model fit
The following research reagent table outlines essential materials for implementing TIDE analysis:
Table 2: Essential Research Reagents for TIDE Analysis
| Reagent/Category | Specific Examples | Function in TIDE Workflow |
|---|---|---|
| Genomic DNA Isolation | BioLine ISOLATE II Genomic DNA Kit, phenol/chloroform extraction [13] | Obtain high-quality genomic DNA from edited and control cells |
| PCR Amplification | BioLine MyTaq Mastermix, high-fidelity polymerases [13] | Amplify target region surrounding nuclease cut site |
| Sequencing Reagents | BigDye Terminator v3.1 (Applied Biosystems) [13] | Generate sequencing traces from PCR amplicons |
| Analysis Software | TIDE web tool (https://tide.nki.nl) [1] [3] | Computational deconvolution of sequencing traces to quantify indels |
Advanced Applications and Methodological Extensions
Specialized Applications in Complex Genomic Contexts
While TIDE was originally developed for standard gene editing applications, its utility has expanded to more complex genomic contexts. In plant genome editing, particularly for challenging multi-copy gene families like the wheat α-gliadins, TIDE and similar tools face limitations due to the presence of highly homologous sequences arranged in tandem across subgenomes [15]. In these contexts, next-generation sequencing amplicon-based approaches with specialized bioinformatic pipelines have proven more effective for comprehensive indel characterization [15]. However, TIDE remains valuable for initial efficiency assessment before moving to more complex analyses.
For therapeutic applications and precise genome editing, the TIDER method extends TIDE's capabilities to quantify template-directed mutations alongside non-templated indels [13]. TIDER requires three sequencing traces instead of two—adding a "reference" trace representing the desired HDR outcome—enabling researchers to distinguish and quantify precise edits from the background of random indels [3] [13]. This capability is particularly valuable for disease modeling and therapeutic development where specific nucleotide changes are required.
Integration with Orthogonal Validation Methods
Best practices in genome editing validation recommend integrating TIDE with orthogonal methods to ensure result reliability. Several studies have demonstrated strong correlation between TIDE analysis and more comprehensive validation approaches:
Next-generation sequencing: While NGS remains the gold standard for comprehensive editing assessment, TIDE shows reasonable correlation for efficiency estimation while being more accessible for routine use [4].
T7 Endonuclease I (T7EI) assay: TIDE provides more quantitative and detailed information than the semi-quantitative T7EI assay, which only detects the presence of heteroduplexes without characterizing specific indels [8] [4].
Droplet digital PCR (ddPCR): For absolute quantification of specific edits, ddPCR offers superior precision, though it requires specialized equipment and probe design [8].
A 2025 optimization study for gene knockout in human pluripotent stem cells successfully employed TIDE alongside ICE and T7EI assays, leveraging their complementary strengths to validate editing efficiency across multiple sgRNAs and target sites [16]. This integrated approach provides a balance of throughput, cost, and analytical depth suitable for different stages of the research pipeline.
The TIDE methodology represents a significant advancement in making quantitative genome editing assessment accessible to broad research communities. By transforming standard Sanger sequencing traces into detailed indel spectra through sophisticated computational decomposition, TIDE bridges the gap between simple presence/absence assays and comprehensive next-generation sequencing. As the field of genome editing continues to evolve toward more complex applications—including base editing, prime editing, and therapeutic development—the fundamental principles underlying TIDE remain relevant for rapid, cost-effective preliminary assessment.
Future methodological developments will likely focus on expanding detection capabilities for larger genomic rearrangements, improving accuracy for complex editing outcomes, and enhancing integration with multi-omics approaches. The recent introduction of tools like PtWAVE for large indel detection demonstrates ongoing innovation in this space [5]. However, TIDE's established workflow, accessibility, and validation across diverse applications ensure its continued relevance in the genome editing toolkit. For researchers pursuing drug development and therapeutic applications, TIDE provides an critical first-pass assessment tool that balances technical depth with practical implementation requirements, enabling more efficient optimization of editing conditions and guide RNA selection before committing to more resource-intensive validation methods.
In the field of genome editing, validating the efficiency of CRISPR-Cas9 systems is a critical step. For years, clonal sequencing—the process of isolating single-cell derived clones and sequencing individual alleles—served as a primary method for assessing editing outcomes. While accurate, this approach is notoriously labor-intensive and slow. The development of the TIDE (Tracking of Indels by Decomposition) assay provided a transformative alternative for analyzing pooled populations of edited cells. This guide objectively compares the performance of the TIDE assay against traditional clonal sequencing, focusing on the key practical advantages of speed, cost, and accessibility that make TIDE a superior choice for initial screening and optimization in most research contexts.
Key Advantages of TIDE vs. Clonal Sequencing
The following table summarizes the core advantages of using the TIDE assay over traditional clonal sequencing for the initial assessment of CRISPR editing efficiency.
Table 1: Core Advantages of TIDE Assay Over Clonal Sequencing
| Feature | TIDE Assay | Traditional Clonal Sequencing | Practical Implication for Researchers |
|---|---|---|---|
| Speed & Workflow | 1-2 days from DNA to result [1] [5] | Several weeks [5] | Drastically faster feedback for guide RNA validation and experimental iteration. |
| Labor Input | Low; avoids clonal isolation [5] [17] | High; requires clonal expansion and picking [5] [17] | Frees up significant researcher time and reduces manual cell culture work. |
| Cost | Low (relies on Sanger sequencing) [4] [17] | High (cloning, transformation, multiple sequencing reactions) [5] | More cost-effective for screening multiple guide RNAs or experimental conditions. |
| Technical Accessibility | High; uses standard lab equipment (PCR, Sanger sequencer) [1] [17] | Moderate; requires proficiency in cloning techniques [5] | Accessible to a broader range of labs without specialized expertise in molecular cloning. |
| Data Output | Quantifies overall editing efficiency and identifies predominant indel spectra [1] [7] | Provides exact sequence of each individual allele [5] [17] | Ideal for assessing overall nuclease activity rather than cataloging every single edit. |
Experimental Data and Benchmarking
The practical advantages of TIDE are supported by experimental data that benchmarks its performance against sequencing-based methods.
Quantitative Accuracy Compared to Next-Generation Sequencing
While Next-Generation Sequencing (NGS) is considered the "gold standard" for comprehensive editing analysis [4] [18], TIDE provides a highly accurate estimate of overall editing efficiency that is sufficient for most validation purposes. One study noted that TIDE, along with other Sanger-based tools, predicts editing efficiencies that are highly similar to those observed with targeted NGS for pools of cells [7]. This correlation makes TIDE a reliable and cost-effective substitute for NGS when deep sequence-level detail is not required.
Superior Quantitative Performance Over Enzymatic Assays
TIDE offers a significant advantage over non-sequencing based methods like the T7 Endonuclease I (T7E1) assay. While T7E1 is cheap and fast, it is only semi-quantitative and has a low dynamic range, often failing to accurately reflect the true editing efficiency observed by sequencing methods [4] [7] [8]. Research has shown that T7E1 can dramatically underestimate the activity of highly efficient sgRNAs and fail to detect low-activity ones altogether [7]. In contrast, TIDE provides a more quantitative and sensitive measurement of indel frequencies.
Standard TIDE Assay Protocol
The following workflow outlines the standard experimental procedure for the TIDE assay, highlighting its straightforward nature compared to clonal sequencing.
Diagram 1: TIDE Assay Workflow
Detailed Experimental Methodology
- Perform CRISPR Editing and Collect Samples: Transfert or transduce cells with CRISPR-Cas9 components (e.g., plasmid, RNP). Include a negative control (e.g., cells without Cas9 or with a non-targeting gRNA). Harvest genomic DNA from the edited pool and the control pool typically 3-4 days after transfection [7] [8].
- PCR Amplification: Design primers to amplify a genomic region of ~500-700 bp surrounding the CRISPR target site. The projected Cas9 cut site should be located preferably ~200 bp downstream from the sequencing start site to ensure high-quality sequence data for analysis [1]. Perform PCR using high-fidelity polymerase on both the edited and control DNA samples.
- Sanger Sequencing: Purify the PCR products and submit them for Sanger sequencing using one of the PCR primers. The output will be chromatogram files (e.g., in .ab1 or .scf format) for both the control and edited samples [1].
- Computational Analysis with TIDE:
- Access the TIDE web tool (https://apps.datacurators.nl/tide/).
- Input the sgRNA target sequence (20nt, excluding the PAM).
- Upload the control and edited sample sequencing files.
- Set the parameters, typically using the default settings (e.g., decomposition window, indel size range of 10 bp, p-value threshold < 0.001). The tool assumes the double-strand break is induced between nucleotides 17 and 18 of the sgRNA sequence [1].
- Run the analysis. The tool decomposes the complex sequencing trace from the edited pool and provides an indel efficiency report and a spectrum of the predominant edits.
Research Reagent Solutions
The following table lists the key materials and resources required to perform the TIDE assay.
Table 2: Essential Reagents and Tools for the TIDE Assay
| Item | Function / Description | Example / Note |
|---|---|---|
| CRISPR-Cas9 System | To induce targeted double-strand breaks. | Can be delivered as plasmid, mRNA, or ribonucleoprotein (RNP) complexes [17]. |
| Cell Culture Reagents | For maintaining and transfecting the cell line of interest. | Specific media, transfection reagent. |
| Genomic DNA Extraction Kit | To isolate high-quality DNA from the pooled edited cells and control cells. | Standard commercial kits are suitable. |
| PCR Reagents | To amplify the target locus from genomic DNA. | High-fidelity DNA polymerase, dNTPs, specific primers [1]. |
| Sanger Sequencing Service | To generate sequence chromatograms of the PCR amplicons. | Core facility or commercial service; requires .ab1 or .scf file output [1]. |
| TIDE Web Tool | The online software that deconvolutes the sequencing traces to quantify indel frequency and spectrum. | Freely available for academic, non-commercial use [1]. |
The TIDE assay presents a compelling case for replacing clonal sequencing as the first choice for validating CRISPR genome editing efficiency. Its principal advantages in speed (days versus weeks), cost-effectiveness (leveraging Sanger sequencing), and accessibility (a simple protocol with a free web tool) enable researchers to streamline their workflows and accelerate experimental timelines. While clonal sequencing remains necessary for confirming the exact sequence of homozygous edits in individual cell lines, the TIDE assay is objectively superior for the rapid and quantitative assessment of editing in bulk cell populations, making it an indispensable tool in the modern genome editor's toolkit.
How to Perform a TIDE Assay: A Step-by-Step Protocol from Lab to Analysis
The foundation of any successful TIDE (Tracking of Indels by DEcomposition) analysis rests upon the quality of the initial sample preparation. As a method that quantifies the spectrum and frequency of small insertions and deletions (indels) generated by genome editing tools, TIDE requires high-quality input data derived from precise PCR amplification and Sanger sequencing [1] [9]. This first step is critical; the accuracy of the decomposition algorithm is wholly dependent on clean sequence chromatograms from both control and edited DNA samples. This guide details the essential requirements for sample preparation and objectively compares TIDE's performance and protocol against other common genome editing analysis methods.
PCR Requirements and Best Practices
Proper PCR amplification of the target locus is the first crucial step in generating material for TIDE analysis.
Primer Design and Specificity
- Design Criteria: PCR primers should be specific to the target sequence, free of internal secondary structure, and devoid of polybase sequences or repeating motifs [19]. Primer pairs should have compatible melting temperatures (within 5°C) and approximately 50% GC content.
- Amplicon Length and Quality: It is advised to sequence a DNA stretch of approximately 700 base pairs enclosing the designed editing site [1]. The PCR product must appear as a single, specific band on an agarose gel. Multiple bands indicate non-specific amplification, which will lead to mixed template sequencing and poor-quality results [20] [19].
PCR Product Cleanup
After amplification, the PCR product must be purified to remove residual reagents such as excess primers, nucleotides, and enzymes that can interfere with the subsequent sequencing reaction [20].
- Purpose: Removal of excess PCR primers is essential because both primers will work in a sequencing reaction and produce a mixed sequence read [20].
- Methods: Several methods are available, including:
- Enzymatic purification: Using shrimp alkaline phosphatase (SAP) and Exonuclease I (Exo I) to degrade nucleotides and single-stranded DNA primers [19].
- Column-based purification: Kits such as the Qiaquick (Qiagen) PCR ISOLATE II PCR and Gel Kit (Bioline) are effective [20] [9].
- Critical Note: If multiple PCR products are present, only gel purification will isolate the desired product; column purification or enzymatic cleanup will not [19].
Template Quantification
Accurate quantification of the purified DNA is vital for a successful sequencing reaction.
- Challenge: Quantifying a PCR sample before purification will yield an incorrect concentration due to the presence of primers and nucleotides [20]. Spectrophotometer readings (e.g., Nanodrop) are only accurate when the A260 is between 0.1 and 0.8. Samples above this range must be diluted and re-measured [20].
- Recommendation for PCR Products: For purified PCR products, use a fluorometer for accurate quantitation or estimate concentration based on band intensity relative to mass standards on an agarose gel [21].
Sanger Sequencing Submission Guidelines
Following PCR cleanup, the sample is prepared for Sanger sequencing.
Sample Preparation for Sequencing
Adherence to service provider guidelines ensures high-quality sequence traces. The table below summarizes typical template and primer requirements for Sanger sequencing services.
Table 1: Sanger Sequencing Sample Submission Guidelines
| DNA Type | DNA Length | Template Concentration | Template Total Mass | Primer Concentration |
|---|---|---|---|---|
| Plasmids | < 6 kb | ~50 ng/µL | ~500 ng | 5 µM [21] |
| Plasmids | 6 – 10 kb | ~80 ng/µL | ~800 ng | 5 µM [21] |
| Plasmids | > 10 kb | ~100 ng/µL | ~1000 ng | 5 µM [21] |
| Purified PCR Products | < 500 bp | ~1 ng/µL | ~10 ng | 5 µM [21] |
| Purified PCR Products | 500 – 1000 bp | ~2 ng/µL | ~20 ng | 5 µM [21] |
| Purified PCR Products | 1000 – 2000 bp | ~4 ng/µL | ~40 ng | 5 µM [21] |
Sequencing Reaction Setup
The sequencing itself is typically performed with 4 µL of BigDye terminator mix and 5 pM of a single sequencing primer in a 20 µL reaction volume [9]. The sequencing primer can be one of the PCR primers or a nested primer. The use of universal-tailed primers (e.g., M13 tails) can standardize and simplify the sequencing setup for large projects [19].
Comparative Analysis of Genome Editing Assessment Methods
While TIDE is a powerful tool, researchers should be aware of how it compares to other available methods for assessing genome editing efficiency. The following table provides a high-level comparison of key techniques.
Table 2: Comparison of Genome Editing Efficiency Assessment Methods
| Method | Principle | Throughput | Cost | Quantitative Capability | Information on Indel Spectrum |
|---|---|---|---|---|---|
| TIDE | Decomposition of Sanger sequencing traces [1] | Medium | Low | Accurate quantification of major indels [10] | Identifies predominant indels and their frequencies [1] |
| ICE (Inference of CRISPR Edits) | Decomposition of Sanger sequencing traces [4] | Medium | Low | Accurate (comparable to NGS) [4] | Identifies indels and their distributions; can detect large indels [4] |
| NGS (Next-Generation Sequencing) | Deep sequencing of amplicons [4] | High | High | Highly accurate and sensitive [4] | Most comprehensive; reveals full heterogeneity [10] |
| T7 Endonuclease I (T7E1) Assay | Cleavage of heteroduplex DNA [8] | High | Very Low | Semi-quantitative [8] | No sequence information [4] |
| ddPCR | Fluorescent probe-based detection [8] | Medium | Medium | Highly precise and quantitative [8] | Limited to predefined edits |
Supporting Experimental Data from Comparative Studies
Independent studies have systematically evaluated the performance of these computational tools.
- A 2024 study compared TIDE, ICE, DECODR, and SeqScreener using artificial sequencing templates with predetermined indels. The study found that while these tools could estimate indel frequency with acceptable accuracy for simple indels, the estimated values became more variable with complex indels. DECODR provided the most accurate estimations for most samples, though TIDER (a TIDE-based tool) performed best for assessing knock-in efficiency [10].
- A 2025 comparative analysis highlighted that TIDE and ICE offer more quantitative analysis than T7E1 assays but noted that their accuracy heavily relies on the quality of PCR amplification and sequencing [8].
The Scientist's Toolkit: Essential Research Reagents
A successful TIDE experiment requires several key reagents and materials, as detailed below.
Table 3: Essential Reagents and Materials for TIDE Analysis
| Item | Function/Description | Example Products/Notes |
|---|---|---|
| High-Fidelity DNA Polymerase | Amplifies the genomic target region with high accuracy. | KOD One PCR Master Mix [10], Q5 Hot Start High-Fidelity Master Mix [8] |
| PCR Purification Kit | Removes excess primers, dNTPs, and enzymes post-amplification. | Qiaquick (Qiagen), ISOLATE II PCR and Gel Kit (Bioline) [20] [9] |
| Sanger Sequencing Kit | Performs the cycle sequencing reaction with fluorescent dye-terminators. | BigDye Terminator v3.1 (Applied Biosystems) [9] |
| Genomic DNA Isolation Kit | Extracts high-quality genomic DNA from edited cells. | ISOLATE II Genomic DNA Kit (Bioline) [9] |
| TIDE Web Tool | The online software that decomposes sequencing chromatograms to quantify indels. | Available at http://shinyapps.datacurators.nl/tide/ [1] [8] |
Experimental Workflow for TIDE Analysis
The following diagram illustrates the end-to-end process from cell editing to data analysis.
TIDE Assay Experimental Workflow
Detailed TIDE Protocol from Cited Literature
The methodology for a TIDE experiment, as described in the foundational paper, involves the following steps [9]:
- Cell Transfection and DNA Extraction: Transfert cells with the CRISPR-Cas9 plasmid (e.g., PX330). After 3 days, isolate genomic DNA from approximately 1 million cells using a commercial kit (e.g., ISOLATE II Genomic DNA Kit, Bioline).
- PCR Amplification: Perform PCR with 50 ng of genomic DNA using a proofreading polymerase (e.g., MyTaq Red mix, Bioline). Use primers that span the target site, generating a product that encompasses the cut site. Purify the resulting PCR product (e.g., with PCR ISOLATE II PCR and Gel Kit).
- Sanger Sequencing: Submit ~30 ng of purified PCR product for Sanger sequencing using the BigDye Terminator v3.1 kit and 5 pM of a single sequencing primer.
- TIDE Analysis:
- Input: Upload the sequencing chromatogram files (
.ab1or.scf) for both the control and edited samples to the TIDE web tool [1]. - sgRNA Sequence: Provide the 20nt sgRNA sequence (without the PAM) [1].
- Parameters: The tool assumes the double-strand break is induced between nucleotides 17 and 18 of the sgRNA sequence. Default analysis parameters (e.g., indel size range of 10, p-value threshold < 0.001) are typically sufficient, but advanced settings can be adjusted [1].
- Output: The software provides an indel spectrum plot, quantification of indel frequencies, an R² value for goodness of fit, and a quality control plot showing the aberrant sequence signal [1].
- Input: Upload the sequencing chromatogram files (
Meticulous attention to the PCR and Sanger sequencing requirements outlined in this guide is paramount for generating reliable data for TIDE analysis. The method provides a robust and cost-effective solution for quantifying genome editing efficiency, particularly suited for labs without access to NGS. When selecting an analysis method, researchers must weigh the need for detailed sequence information against factors such as throughput, cost, and technical complexity. For many applications, TIDE strikes an effective balance, providing quantitative data on the predominant indels from standard Sanger sequencing traces.
Input Requirements for TIDE Analysis
The TIDE assay requires two specific types of input data for successful decomposition of editing outcomes [1] [9].
Guide RNA Sequence Specification
- Sequence: A 20-nucleotide ('5-'3) DNA character string representing the sgRNA guide sequence immediately upstream of the PAM sequence (PAM not included) [1].
- Break Site: TIDE assumes that a double-strand break is induced between nucleotides 17 and 18 in the provided sgRNA sequence [1].
- Formatting: The software automatically removes numbers and other invalid (non-IUPAC) DNA characters from the input [1].
Sequencing File Formats
TIDE accepts standard capillary sequencing file formats [1] [22]:
| File Format | Description | Developer/Type | Key Features for TIDE |
|---|---|---|---|
| .ab1 | ABI sequencer output file; raw trace file | Applied Biosystems [22] | Contains raw data, base calls, quality scores, and electropherogram trace data [22]. |
| .scf | Standard Chromatogram Format | Open-source format [22] | Carries the same trace information as .ab1 files and is universally readable [22]. |
Comparative Analysis: TIDE vs. Alternative Methods
TIDE provides distinct advantages over traditional enzyme-based assays and high-throughput sequencing for initial efficiency screening [9] [13].
Quantitative Comparison of Genome Editing Assessment Methods
| Method | Time Required | Cost | Labor Intensity | Information Detail | Primary Use Case |
|---|---|---|---|---|---|
| TIDE | ~1 day [13] | Low [9] | Low [9] | Identifies and quantifies major indel types and their frequencies [9] [13]. | Rapid testing and optimization of editing conditions [1]. |
| T7E1/Surveyor | ~1 day [9] | Low [9] | Low [9] | Semi-quantitative; does not provide information on the nature or diversity of mutations [9]. | Basic, low-detail efficacy confirmation [9]. |
| Cloning & Sanger | Several days to weeks [9] | Medium [9] | High [9] | Detailed; identifies all sequence variants but is low-throughput [9]. | In-depth analysis of a small number of samples [9]. |
| High-Throughput Sequencing | Several weeks [9] | High [9] | Medium (post-processing) | Highly detailed; comprehensive profile of all indels and their frequencies [9]. | Final, comprehensive validation and publication data [9]. |
Experimental Protocol for TIDE Sample Preparation
The following protocol is adapted for TIDE analysis to ensure high-quality results [13].
PCR Amplification
- Primer Design: Design primers that flank the expected break site, aiming to amplify a DNA stretch of 500–1500 bp. The projected break site should be located preferably ~200 bp downstream from the sequencing primer binding site [1] [13].
- PCR Reaction:
- Genomic DNA: ~50 ng (isolated from a minimum of 1000 cells 3 days post-transfection) [9] [13].
- Primers: 2 µL of each primer (from a 10 µM stock) [13].
- Master Mix: 25 µL of a 2x pre-mix (e.g., BioLine MyTaq Red mix) [13].
- PCR Program: Initial denaturation at 95°C for 1 min; 25-30 cycles of 95°C for 15s, 55-58°C for 15s, 72°C for 10s-1min; final extension at 72°C for 1 min [13].
- Purification: Purify the PCR product using a kit (e.g., BioLine ISOLATE II PCR and Gel Kit) [13].
Sanger Sequencing
- Sequencing Reaction:
- Data Export: Analyze samples on a capillary sequencer (e.g., Applied Biosystems 3730xl) and save the sequence trace files in .ab1 or .scf format [1] [13].
Workflow from Sample to Analysis
The Scientist's Toolkit: Essential Research Reagents
| Reagent/Item | Function in TIDE Workflow |
|---|---|
| Programmable Nuclease (e.g., CRISPR-Cas9, TALENs, ZFNs) | Induces a targeted double-strand break at the genomic locus of interest [9]. |
| sgRNA Expression Plasmid (e.g., PX330 for CRISPR) | Directs the nuclease to the specific target site upstream of the PAM sequence [9]. |
| Genomic DNA Isolation Kit (e.g., BioLine ISOLATE II) | Purifies high-quality genomic DNA from a pool of transfected cells for PCR amplification [13]. |
| PCR Master Mix (e.g., BioLine MyTaq Red) | Amplifies the target genomic region surrounding the nuclease break site [13]. |
| PCR Purification Kit | Removes primers, enzymes, and salts to ensure a clean template for Sanger sequencing [13]. |
| BigDye Terminator Kit (v3.1) | Used in the cycle-sequencing reaction to generate fluorescently labeled DNA fragments for capillary electrophoresis [13]. |
Tracking of Indels by Decomposition (TIDE) is a computational method that rapidly quantifies the efficiency and spectrum of small insertions and deletions (indels) generated by genome editing tools like CRISPR-Cas9 in a pool of cells [1]. By decomposing Sanger sequencing chromatograms from edited samples, TIDE provides researchers with a comprehensive profile of editing outcomes without requiring costly next-generation sequencing [1] [10].
The accuracy of TIDE analysis heavily depends on proper configuration of two fundamental parameters: the alignment window and decomposition window [1]. These parameters define which segments of the sequencing trace data are used for aligning control and test sequences and for computational decomposition to identify indels. Correct configuration ensures precise quantification of editing efficiency, while improper settings can lead to inaccurate indel detection and frequency estimation [1] [10].
Defining Alignment and Decomposition Windows
Alignment Window: Purpose and Configuration
The alignment window specifies the sequence segment used to align the control (unedited) and test (edited) sample sequencing traces [1]. This alignment is crucial for detecting and correcting any offset between the two sequencing reads before decomposition analysis.
Default Settings and Adjustment Guidelines:
- Left boundary: Default is 100 base pairs from the sequencing start [1]
- Right boundary: Automatically set at the break site minus 10 bp [1]
- Adjustment scenarios: Reduce the left boundary when sequence reads are shorter than the recommended 700 bp or when the break site is closer to the sequencing start [1]
The region upstream of the break site within this window enables TIDE to properly align sequencing data between control and test samples, establishing a reference framework for subsequent decomposition analysis [1].
Decomposition Window: Purpose and Configuration
The decomposition window defines the sequence segment used for the actual computational decomposition that identifies and quantifies different indels in the edited sample [1].
Default Settings and Adjustment Guidelines:
- Left boundary: Set at maximum indel size plus 5 bp downstream of the break site [1]
- Right boundary: Set at maximum indel size plus 5 bp before the end of the shortest sequence read [1]
- Default configuration: Uses the largest window possible for the uploaded sequences [1]
A larger decomposition window generally provides more robust estimation of mutations, as it incorporates more sequence information for the decomposition algorithm [1].
Visualizing the TIDE Analysis Workflow
The following diagram illustrates the complete TIDE analysis process, highlighting the role of alignment and decomposition windows:
TIDE Analysis Workflow and Parameters
Comparative Performance of Sanger-Based Editing Analysis Tools
Tool Comparison Framework and Methodology
Recent studies have systematically evaluated the performance of TIDE against other Sanger sequencing-based genome editing analysis tools using artificial sequencing templates with predetermined indels [10]. This approach enables quantitative assessment of accuracy by comparing tool outputs against known indel frequencies and types.
Experimental Methodology:
- Template preparation: Cloned indels induced by CRISPR-Cas9 or CRISPR-Cas12a at various zebrafish gene loci were mixed in predetermined ratios [10]
- Data analysis: Sanger sequencing trace data from various combinations of known indels were processed through multiple computational tools [10]
- Performance metrics: Tools were evaluated on indel frequency estimation accuracy, ability to deconvolute complex indel sequences, and performance across different editing scenarios [10]
Quantitative Performance Comparison
The table below summarizes the comparative performance of major Sanger-based editing analysis tools based on controlled experimental data:
Table 1: Performance Comparison of Sanger-Based Genome Editing Analysis Tools
| Tool | Indel Frequency Accuracy | Complex Indel Handling | Key Strengths | Key Limitations |
|---|---|---|---|---|
| TIDE | Variable with complex patterns [10] | Limited for large (>50 bp) indels [5] | Rapid analysis, user-friendly web interface [1] | Limited detection range for large indels [5] |
| ICE | High correlation with NGS (R² = 0.96) [4] | Better for large indels than TIDE [4] | Comparable to NGS, detects unexpected editing outcomes [4] | Web-based only, requires internet access |
| DECODR | Most accurate for majority of samples [10] | Improved for complex indels [10] | Accurate sequence deconvolution [10] | Less user-friendly interface |
| PtWAVE | Superior for large deletions [5] | Excellent for deletions up to 200 bp [5] | Wide detection range, high sensitivity [5] | Newer tool, less established |
Performance Across Different Editing Scenarios
The same study revealed that all tools showed reasonable accuracy with simple indels containing few base changes, but performance diverged significantly with more complex editing scenarios [10]:
Table 2: Performance Across Different Editing Scenarios
| Editing Scenario | Best Performing Tool | Key Findings |
|---|---|---|
| Simple indels (few base changes) | All tools adequate [10] | Minimal variation in estimated values among tools |
| Complex indels/multiple variants | DECODR [10] | Significant variability in estimates among tools |
| Low or high indel frequency ranges | DECODR [10] | Greater estimation accuracy at frequency extremes |
| Large deletions (>50 bp) | PtWAVE [5] | Superior accuracy and sensitivity for large deletions |
| Knock-in efficiency estimation | TIDER (TIDE-based) [10] | Specialized functionality for knock-in analysis |
Experimental Protocols for Method Comparison
Plasmid Mixture Model for Tool Validation
A established experimental approach for validating editing analysis tools involves creating plasmid mixtures with known ratios of wild-type and edited sequences:
Protocol:
- Model construction: Two plasmids with wild-type and edited sequences (e.g., 3-bp difference) are mixed in varying ratios from 0% to 100% [8]
- PCR amplification: Primers flanking the target site are used to amplify the region using high-fidelity polymerase [8]
- Sequencing: Sanger sequencing is performed on the mixed plasmid samples [8]
- Tool analysis: Resulting sequencing files are analyzed with different tools and compared to known mixing ratios [8]
This approach provides a controlled system for evaluating tool accuracy independent of biological variables.
Quality Control Metrics for TIDE Analysis
TIDE provides built-in quality metrics to evaluate analysis reliability:
Key Quality Indicators:
- R² value: Should be >0.9 for decomposition results, indicating good fit [1]
- Aberrant sequence signal: Should be <10% before breaksite in both control and test samples [1]
- Control sample profile: Should show low, equally distributed aberrant sequence signal [1]
- Test sample profile: Should show low signal before breaksite and higher signal downstream [1]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for TIDE Analysis
| Reagent/Software | Function/Purpose | Specifications |
|---|---|---|
| Q5 Hot Start High-Fidelity Master Mix | PCR amplification of target locus | High-fidelity polymerase for accurate amplification [8] |
| T7 Endonuclease I | Alternative mismatch detection assay | Cleaves heteroduplex DNA at mismatch sites [8] |
| Gel and PCR Clean-Up Kit | Purification of PCR products | Removes primers and enzymes before sequencing [8] |
| Sanger Sequencing Services | Capillary sequencing of amplicons | Provides .ab1 chromatogram files for TIDE analysis [8] |
| TIDE Web Tool | Primary analysis platform | Decomposes sequencing traces to quantify indels [1] |
| ICE (Inference of CRISPR Edits) | Comparative analysis tool | Provides alternative quantification method [4] |
Impact of Window Configuration on Data Interpretation
Consequences of Improper Window Settings
Incorrect configuration of alignment and decomposition windows can significantly impact TIDE results:
- Overly restrictive decomposition windows may miss larger indels or provide insufficient sequence context for accurate decomposition [1]
- Excessively large decomposition windows that include poor-quality sequence regions can introduce noise and reduce accuracy [5]
- Improper alignment windows may fail to correctly align control and test sequences, leading to erroneous decomposition [1]
Optimization Guidelines for Different Applications
For Standard Editing Experiments:
- Use default window settings initially [1]
- Verify quality metrics (R² > 0.9, aberrant signal <10%) [1]
- Sequence opposite strand to confirm results [1]
For Experiments with Large Expected Indels:
- Increase indel size range parameter beyond default 10 bp [1]
- Consider using specialized tools like PtWAVE for deletions up to 200 bp [5]
- Ensure decomposition window accommodates expected indel sizes [5]
For Low-Quality Sequencing Data:
- Adjust alignment window to avoid poor-quality regions [1]
- Reduce decomposition window to exclude areas with high aberrant signals [1]
- Consider re-sequencing samples if quality metrics cannot be met [1]
Proper configuration of alignment and decomposition windows remains essential for obtaining accurate, reliable results from TIDE analysis, while understanding tool limitations helps researchers select the most appropriate method for their specific genome editing application.
This guide details the interpretation of results from the Tracking of Indels by Decomposition (TIDE) assay, objectively compares its performance against other CRISPR analysis methods, and provides supporting experimental data and protocols.
Core Components of TIDE Results Interpretation
The TIDE web tool generates several key outputs that require careful interpretation to accurately assess the outcome of a genome editing experiment.
Indel Spectrum and Frequencies: The primary output of TIDE is a comprehensive profile of all insertions and deletions (indels) in the edited sample. The algorithm uses quantitative sequence trace data from capillary sequencing to decompose the composite sequence trace from the edited sample into its constituent indel sequences. It reports the identity of each detected indel and its frequency within the pool of cells. The frequency is presented as a percentage, representing the relative abundance of each specific indel mutation generated by the genome editing tool. The statistical significance of the detection of each indel is calculated, and a default P-value threshold of < 0.001 is applied to determine significance [1].
R² Value (Goodness of Fit): The R² value is calculated as a measure of the goodness of fit of the decomposition model. It indicates how well the combination of trace models (the predicted indels) explains the composite sequence trace obtained from the experimental sample. As a rule of thumb, an R² value of > 0.9 is recommended for a reliable decomposition result. A lower R² value suggests that the model does not fully account for all the sequence variation in the edited sample, which could be due to poor sequence quality or the presence of complex mutations that the algorithm cannot deconvolute [1].
Aberrant Sequence Signal Plot: This quality control plot displays the percentage of aberrant nucleotide signals along the sequence trace for both the control and experimental samples. A value of 0% at a position indicates no difference from the control sequence, while 100% indicates the expected nucleotide was not detected at all. For a good quality experiment, the control sample should show a low and equally distributed aberrant sequence signal. The test sample should show a low signal before the breaksite and a consistently elevated signal downstream of the breaksite, indicating the region where sequences deviate due to indels [1].
+1 Insertion Plot: For the common occurrence of a single base pair insertion (+1 insertion), TIDE provides a specific plot that estimates the base composition of this insertion [1].
Performance Comparison with Alternative Methods
The following table summarizes a direct comparison of TIDE with other commonly used methods for assessing CRISPR-Cas9 editing efficiency, based on recent comparative studies.
Table 1: Comparative Analysis of CRISPR-Cas9 Editing Assessment Methods
| Method | Principle | Key Performance Metrics | Detection Range / Limitations | Best Use Cases |
|---|---|---|---|---|
| TIDE [1] [8] [2] | Deconvolution of Sanger sequencing traces via non-negative linear regression. | - High correlation with NGS for simple indels [4].- Accuracy decreases with complex indel mixtures or very low/high editing frequencies [2].- Limited in detecting large indels (>50 bp) [5]. | - Default indel size range is 10 bp [1].- Struggles with large deletions and complex knock-in sequences [2] [5]. | Rapid, cost-effective quantification of small indels for routine editing experiments. |
| ICE (Synthego) [4] [6] [2] | Deconvolution of Sanger sequencing traces via LASSO regression. | - Highly comparable to NGS (R² = 0.96) [4].- Can detect larger indels and more complex editing outcomes than TIDE [4].- Shows high variability in indel frequency reporting in somatic in vivo models [6]. | - More capable than TIDE for large insertions/deletions [4].- Performance varies with sample type and complexity [6]. | Users seeking a user-friendly interface with capabilities beyond basic TIDE analysis. |
| T7E1 Assay [8] [7] | Mismatch cleavage of heteroduplex DNA by T7 endonuclease I. | - Semi-quantitative; lacks sensitivity [8].- Often underestimates high editing efficiency and fails to detect low activity [7].- Does not provide sequence-level data [4]. | - Cannot identify specific indel sequences or their individual frequencies [4]. | Low-cost, quick initial screening where precise sequence data is not required. |
| ddPCR [8] | Absolute quantification using fluorescent probes in partitioned samples. | - Highly precise and quantitative for specific, predefined edits [8].- Excellent for discriminating between NHEJ and HDR products [8]. | - Requires prior knowledge of the expected mutation to design probes [8].- Not suitable for discovering unknown indels. | Applications requiring absolute quantification of known allelic modifications. |
| Next-Generation Sequencing (NGS) [4] [6] [7] | High-throughput deep sequencing of the target locus. | - Gold standard for sensitivity and comprehensiveness [4] [6].- Detects the full spectrum of edits, including low-frequency and large complex indels [6]. | - High cost, time-consuming, and requires bioinformatics expertise [4]. | Projects requiring the highest level of detail and accuracy, or with large sample numbers. |
A systematic comparison of computational tools using artificial sequencing templates with predetermined indels revealed that while TIDE, ICE, and DECODR perform well with simple indels of a few base changes, their estimated values become more variable when samples contain complex indels [2]. Another study focusing on somatic in vivo CRISPR/Cas9 tumor models reported "high variability in the reported number, size, and frequency of indels" across TIDE, ICE, and DECODR platforms, particularly when larger indels were present [6].
Table 2: Software Performance in Detecting Large Deletions (>50 bp) [5]
| Software | Underlying Algorithm | Reported Maximum Deletion Detection Capability | Key Finding in Benchmarking |
|---|---|---|---|
| TIDE | Non-negative linear regression (NNLS) | Limited (~50 bp) | Struggles with accurate detection and quantification of large deletions. |
| ICE | Non-negative LASSO regression | Limited (~50 bp) | Shows improved detection over TIDE but still with limitations. |
| DECODR | NNLS (Improved algorithm) | Up to 100 bp | More accurate than TIDE and ICE for a wider range of indels. |
| PtWAVE | Progressive variable selection with NNLS/LASSO | Up to 200 bp | Demonstrated superior accuracy and sensitivity for samples including large deletions. |
Experimental Protocols for Key Comparisons
Protocol: Comparative Analysis of TIDE, ICE, and T7E1 Using Plasmid Mixes
This protocol, adapted from a 2025 comparative study, outlines a method to benchmark the accuracy of different editing efficiency methods using controlled plasmid mixtures [8] [23].
Model Construction
- Construct two plasmids: one containing a wild-type target sequence and another containing a known edited sequence (e.g., with a 3-bp difference).
- Mix the two plasmids in varying ratios (e.g., 0%, 5%, 10%, 25%, 50%, 75%, 100% of the edited plasmid) to simulate a range of known editing frequencies.
PCR Amplification
- Reaction Setup: Use 1 μL of plasmid mixture, 1 μL of each primer, 10.5 μL of RNase-free water, and 12.5 μL of a high-fidelity PCR master mix (e.g., Q5 Hot Start High-Fidelity 2X Master Mix) in a 25 μL final volume.
- Thermocycling:
- Initial Denaturation: 98°C for 30 s.
- 30 Cycles of:
- Denaturation: 98°C for 10 s.
- Annealing: 60°C for 30 s.
- Extension: 72°C for 30 s.
- Final Extension: 72°C for 2 min.
- Verify the PCR product on a 1% agarose gel.
Sanger Sequencing and Analysis with TIDE/ICE
- Purify the PCR products from each mixture ratio.
- Submit the purified amplicons for Sanger sequencing.
- Analyze the resulting sequencing chromatograms (.ab1 files) using the TIDE and ICE web tools.
- For TIDE: Upload the wild-type (0% edited) and each test sample file. Input the gRNA sequence and set the cut site. Use default parameters or an indel size matching the known edit (e.g., 3 bp) [23].
- For ICE: Follow the respective web tool's instructions for data upload and analysis.
- Record the total indel frequency reported by each tool for each known mixture ratio.
T7E1 Assay
- Purify the PCR products from each mixture ratio.
- Heteroduplex Formation: Denature and re-anneal the PCR products using a thermocycler (e.g., 95°C for 10 min, ramp down to 25°C at 0.1°C/s).
- Digestion: Incubate 8 μL of re-annealed product with 1 μL of NEBuffer 2 and 1 μL of T7 Endonuclease I (M0302L, NEB) at 37°C for 30 minutes.
- Analysis: Run the digested products on a 1-2% agarose gel. Image the gel and use densitometry software to quantify the band intensities.
- Calculate Apparent Editing Frequency: Use the formula: % Indel = (1 - sqrt(1 - (a + b)/(A + a + b))) * 100, where A is the intensity of the undigested PCR product band, and a and b are the intensities of the cleaved product bands [7].
Data Comparison
- Plot the editing frequency measured by each method (TIDE, ICE, T7E1) against the known plasmid mixture ratio.
- Calculate the correlation coefficient (R²) and mean absolute error for each method relative to the expected values to objectively assess their accuracy and dynamic range.
Diagram 1: Experimental workflow for benchmarking CRISPR analysis methods using plasmid mixtures.
Protocol: Validating TIDE Results with Targeted NGS
For conclusive validation, especially in complex experimental settings, TIDE results should be compared against the gold standard, NGS [6] [7].
- Sample Preparation: Use the same PCR amplicons that were submitted for Sanger sequencing for NGS library preparation.
- Library Preparation and Sequencing: Prepare a tailed library from the PCR amplicons and perform targeted deep sequencing on a platform such as Illumina MiSeq (e.g., 2 x 250 bp reads).
- NGS Data Analysis: Process the NGS data using a standard bioinformatics pipeline for CRISPR indel analysis (e.g., CRISPResso2) to obtain the true spectrum and frequency of indels.
- Correlation Analysis:
- Compare the total editing efficiency (percentage of reads with indels) reported by NGS with the total indel frequency reported by TIDE.
- Compare the specific indel sequences and their frequencies identified by both methods. TIDE has been shown to be very predictive of overall sgRNA activity in pooled cells, but may deviate in the precise frequency of specific indels, especially in clonal samples or those with large indels [6] [7].
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Materials and Reagents for TIDE Analysis and Validation
| Item | Function / Description | Example Product / Specification |
|---|---|---|
| High-Fidelity DNA Polymerase | Amplifies the target genomic locus with minimal PCR errors, ensuring accurate sequencing. | Q5 Hot Start High-Fidelity Master Mix (NEB) [23] |
| Capillary Sequencing Service | Generates the raw sequencing trace files (.ab1 or .scf) required for TIDE analysis. | Service providing ~700 bp reads, with the breaksite ~200 bp downstream of the primer [1]. |
| T7 Endonuclease I | Enzyme for mismatch cleavage assay; used for quick, low-cost comparison. | T7 Endonuclease I (M0302L, New England Biolabs) [8] [7] |
| Digital Droplet PCR (ddPCR) System | Provides absolute quantification of specific, predefined edits for validation. | System with fluorescent probes designed for wild-type and mutant alleles [8] |
| NGS Library Prep Kit | Prepares amplicon libraries for gold-standard validation using deep sequencing. | Kits for Illumina platforms (e.g., Nextera XT) [7] |
| TIDE Web Tool | The core software for deconvolution of Sanger sequencing traces. | https://tide.nki.nl/ [3] (Free for non-commercial research) |
| TIDER Web Tool | TIDE variant for analyzing templated CRISPR/Cas9 experiments (HDR). | https://tider-calculator.nki.nl/ [24] [3] |
Best Practices for Optimal Sequencing and Data Quality Control
Tracking of Indels by Decomposition (TIDE) is a widely adopted method for quantifying the efficiency and spectrum of small insertions and deletions (indels) generated by genome-editing tools such as CRISPR-Cas9, TALENs, and ZFNs [1] [9]. This assay provides a rapid, cost-effective alternative to next-generation sequencing (NGS) for routine assessment of non-homologous end joining (NHEJ) repair outcomes in pooled cell populations [25]. The core of the TIDE method involves the decomposition of complex Sanger sequencing chromatograms from edited samples, using a control sample to accurately resolve the mixture of induced mutations and determine their individual frequencies [9] [26]. By translating standard Sanger sequencing traces into detailed editing profiles, TIDE eliminates the need for labor-intensive clonal sequencing or semi-quantitative enzymatic assays, offering researchers a practical tool for initial editing validation and guide RNA (gRNA) selection [27] [7].
However, the accuracy of TIDE results is highly dependent on the quality of input materials and sequencing data. Adhering to best practices in sample preparation, sequencing, and data analysis is paramount for generating reliable, reproducible results that accurately reflect the true editing outcomes in a cell population. This guide outlines comprehensive protocols and quality control measures to optimize TIDE sequencing and data interpretation, while objectively comparing its performance against other commonly used validation methods.
Experimental Design and Sample Preparation
A standardized experimental workflow is crucial for obtaining high-quality data for TIDE analysis. The following diagram illustrates the key steps from sample preparation to data interpretation:
Critical Reagents and Materials
The following table details essential research reagent solutions required for successful TIDE analysis:
Table 1: Essential Research Reagent Solutions for TIDE Analysis
| Reagent/Category | Specifications & Functional Role | Protocol Notes |
|---|---|---|
| Genomic DNA Source | ≥1000 transfected cells; Ensures representative sampling of mutation diversity [13]. | Isolate DNA 3-4 days post-transfection using standard kits (e.g., Bioline ISOLATE II) [9]. |
| PCR Primers | Flank target site; Generate ~500-1500 bp amplicon with break site ~200 bp from sequencing start [1] [27]. | Design using standard tools; Verify single, sharp band on agarose gel [13]. |
| PCR Master Mix | High-fidelity polymerase with pre-mixed buffers/dNTPs (e.g., Bioline MyTaq Red mix) [9]. | Reduces amplification bias; 25-30 cycles with annealing at 55-58°C [13]. |
| Sequencing Kit | BigDye Terminator chemistry (e.g., v3.1) [9] [13]. | Uses 100 ng purified PCR product and 5 pmol primer per 20 µl reaction [13]. |
| sgRNA Sequence | 20nt guide sequence (5'-3') immediately upstream of PAM [1]. | Input as DNA string; TIDE assumes cut between nucleotides 17 and 18 [1]. |
Optimal Sequencing Parameters
To generate high-quality sequence traces for TIDE analysis, specific sequencing parameters must be followed:
Amplicon Design: The target region should be amplified to produce a fragment of 500-1500 base pairs enclosing the designed editing site. The projected break site should be positioned approximately 200 bp downstream from the sequencing start site to ensure sufficient sequence coverage both upstream and downstream of the cut site [1] [13]. This configuration provides adequate sequence for proper alignment and decomposition analysis.
Sequencing Reaction: Use purified PCR products (approximately 100 ng) with BigDye Terminator chemistry according to manufacturer protocols. The sequencing primer should be the same as one of the PCR primers used for amplification. Standard cycling conditions include initial denaturation at 96°C for 1 minute, followed by 30 cycles of 96°C for 30 seconds, 50°C for 15 seconds, and 60°C for 4 minutes [13].
File Format: Sequence trace files must be saved in .ab1 (ABIF) or .scf format, as these formats retain the quantitative peak information necessary for TIDE decomposition analysis [1] [9]. Avoid using plain text sequences (FASTA) as they lack the chromatogram data required for accurate quantification.
TIDE Data Analysis and Quality Control
Analysis Workflow and Key Parameters
The TIDE web tool decomposes sequence traces through a multi-step process that aligns samples, identifies indels, and quantifies their frequencies. Understanding this workflow is essential for proper quality control and troubleshooting:
Optimal TIDE analysis requires careful parameter adjustment, particularly through the advanced settings menu:
Alignment Window: This parameter defines the sequence segment used to align the control and test samples. The left boundary is typically set to 100 bp to avoid poor-quality base-calling at the start of the sequence read. The right boundary is automatically set at the break site minus 10 bp [1]. Adjustments may be necessary when long repetitive sequences are present.
Decomposition Window: This critical parameter determines the sequence segment used for decomposition analysis, set downstream of the break site. The default setting is the largest window possible for the uploaded sequences. Generally, larger decomposition windows provide more robust mutation estimation [1]. The left boundary is set at maximum indel size plus 5 bp downstream of the break site, while the right boundary is set at maximum indel size plus 5 bp before the end of the shortest sequence read.
Indel Size Range: This setting controls the maximum size of deletions and insertions to be modeled, with a default value of 10 bp [1]. For experiments where larger indels are expected, this parameter should be increased accordingly to ensure comprehensive detection.
P-value Threshold: This significance cutoff (default p < 0.001) determines which indels are reported as statistically significant [1]. Any value between 0 and 1 is accepted, with lower values providing more stringent filtering of results.
Quality Control Metrics
TIDE provides several quality metrics that researchers must carefully evaluate to ensure data reliability:
Table 2: Key Quality Control Metrics for TIDE Analysis
| QC Metric | Optimal Value/Range | Interpretation & Troubleshooting |
|---|---|---|
| R² Value | > 0.9 [1] [28] | Goodness of fit for decomposition model. Low values indicate poor sequence quality, incorrect parameters, or large indels exceeding set range [28]. |
| Aberrant Sequence Signal (Control) | < 10% before break site [1] | Background noise in control sample. High values indicate poor sequence quality or contamination. |
| Aberrant Sequence Signal (Test) | Low before break site, higher after break site [1] | Expected pattern indicates successful editing. Consistently high signal may indicate general poor quality. |
| Total Editing Efficiency | Theoretically up to 100%, but complex pools may show lower values [28] | Not directly comparable to NGS due to algorithmic differences. Use for relative comparison between samples, not as absolute value. |
Troubleshooting Common Issues
Low R² Values: This common issue can arise from several factors. If large indels (>10 bp) are present, increase the "Indel size range" parameter. For regions with poor local sequence quality, adjust the decomposition window boundaries to exclude problematic areas. Additionally, repetitive sequences in the decomposition window can interfere with analysis and may require boundary adjustment [28].
Inaccurate Efficiency Estimates: TIDE calculations have inherent limitations for complex editing mixtures. In bulk populations with high complexity, TIDE may underestimate true editing efficiency compared to restriction enzyme assays or NGS [28]. Furthermore, the algorithm cannot detect very large deletions (>100 bp) that can originate from CRISPR-Cas9-induced double-strand breaks [1].
Sequence Quality Issues: Poor chromatogram quality with high background noise or incorrectly annotated bases will compromise TIDE analysis. Visually inspect chromatograms for clean peaks with minimal background signals. If quality is poor, consider re-sequencing with adjusted conditions or using a different sequencing primer [1].
Comparative Performance Analysis
TIDE vs. Alternative CRISPR Analysis Methods
When selecting a CRISPR validation method, researchers must consider multiple factors including cost, throughput, and information content. The following table provides a comparative analysis of TIDE against other commonly used methods:
Table 3: Comprehensive Comparison of CRISPR Genome Editing Analysis Methods
| Method | Principle | Detection Limit | Information Content | Cost & Throughput | Key Advantages | Key Limitations |
|---|---|---|---|---|---|---|
| TIDE | Decomposition of Sanger sequence traces [9] | ~1-5% [4] | Identifies predominant indels and their frequencies [1] | Low cost, rapid (1-2 days) [25] | Simple workflow, quantitative, provides indel sequence information [27] | Underestimates efficiency in complex pools [28], cannot detect large deletions [1] |
| T7 Endonuclease I (T7E1) | Enzyme cleavage of mismatched DNA heteroduplexes [4] | ~5-10% [7] | Semi-quantitative editing frequency only [4] | Very low cost, rapid (1 day) [4] | Simple, fast, no sequencing required [4] | No indel identity, inaccurate for high efficiency editing [7] |
| Next-Generation Sequencing (NGS) | High-throughput sequencing of amplicons [4] | ~0.1-1% [4] | Comprehensive indel spectrum and frequency [4] | High cost, slow (weeks) [25] | Gold standard, highest sensitivity and accuracy [4] | Expensive, requires bioinformatics expertise [4] |
| ICE (Inference of CRISPR Edits) | Advanced decomposition of Sanger sequences [4] | ~1-5% [4] | Identifies indels and their frequencies, detects large indels [4] | Low cost, rapid (1-2 days) [4] | Comparable to NGS (R² = 0.96), user-friendly interface [4] | Proprietary algorithm (Synthego) |
Experimental Validation Data
Independent studies have validated TIDE performance against established methods. A comprehensive survey published in Scientific Reports compared editing efficiencies predicted by TIDE with targeted NGS for both cellular pools and single-cell derived clones [7]. The study found that TIDE, along with IDAA (Indel Detection by Amplicon Analysis), predicted similar editing efficiencies to NGS for pools of edited cells [7]. However, the study also highlighted that TIDE can miscall alleles in edited clones, deviating by more than 10% from NGS-predicted indel frequencies in 50% of clones tested [7].
When compared to the T7E1 assay, TIDE demonstrates superior accuracy and information content. While T7E1 often incorrectly reports sgRNA activities due to its low dynamic range and dependence on DNA heteroduplex formation, TIDE provides precise quantification and identification of specific indels [7]. For example, sgRNAs with apparently similar activity in the T7E1 assay (~28%) showed dramatically different editing efficiencies when analyzed by NGS (40% vs. 92%), while TIDE accurately reflected these differences [7].
User experiences from scientific forums further support TIDE's utility in real-world applications. One researcher reported: "If I TOPO clone and sequence the alleles from single clones, I get exactly the indels detected by TIDE" [28]. However, the same discussion noted that "for a gRNA that give me about 100% indels as determined by the loss of a restriction site sitting on the target, TIDE calculated 70% mutagenesis only," highlighting the potential for underestimation in highly complex pools [28].
The TIDE assay represents a balanced solution for researchers seeking to validate CRISPR genome editing experiments, offering an optimal compromise between information content, cost, and throughput when compared to alternative methods. By following the best practices outlined in this guide—including careful experimental design, optimal sequencing parameters, rigorous quality control, and appropriate parameter adjustment—researchers can maximize the reliability and interpretability of their TIDE results. While TIDE has limitations in detecting very large deletions and may underestimate efficiency in highly complex pools, its ability to provide rapid, quantitative indel profiling makes it an invaluable tool for the initial assessment and optimization of genome editing experiments. For applications requiring absolute quantification or detection of large structural variations, orthogonal validation with NGS may be necessary, but for most routine editing assessments, TIDE provides sufficient accuracy with substantially reduced resource requirements.
Solving Common TIDE Problems and Optimizing Your Assay Parameters
In the field of genome editing, the TIDE (Tracking of Indels by Decomposition) assay has become a widely adopted method for quantifying the efficiency and spectrum of CRISPR-induced mutations due to its cost-effectiveness and simplicity [2] [9]. However, the accuracy of its results is profoundly dependent on the quality of the input Sanger sequencing data. Poor sequence traces can lead to inaccurate estimations of indel frequency and misinterpretation of editing outcomes [1]. This guide provides a systematic, evidence-based comparison of how data quality impacts TIDE analysis relative to other modern methods and offers concrete strategies for optimization.
The Critical Link Between Data Quality and Assay Performance
The TIDE algorithm functions by decomposing complex Sanger sequencing traces from edited samples, which represent a mixture of different DNA sequences, into their constituent indel components. It does this by comparing the test sample trace to a control (wild-type) trace [9]. The quality of the sequencing trace data is paramount for this decomposition process to succeed.
Key quality parameters for TIDE, as outlined by its developers, include [1]:
- An average aberrant sequence signal strength before the breaksite of <10% for both control and test samples.
- A goodness-of-fit R² value > 0.9 for the decomposition result.
- A clear increase in the aberrant sequence signal downstream of the expected cut site in the test sample compared to the control.
Sequencing traces with high background noise, low signal intensity, or excessive peak overlapping compromise the algorithm's ability to resolve the complex mixture of sequences, leading to unreliable indel quantification [29].
Comparative Performance of TIDE and Alternative Methods
A 2024 systematic comparison of computational tools for Sanger sequencing analysis, including TIDE, ICE, DECODR, and SeqScreener, revealed that while all tools can estimate indel frequency with reasonable accuracy for simple indels, their performance diverges with complex editing patterns or suboptimal data [2].
Table 1: Tool Performance with Variable Data and Edit Complexity
| Tool | Performance with Simple Indels (few base changes) | Performance with Complex Indels/Knock-ins | Key Limitation |
|---|---|---|---|
| TIDE | Acceptable accuracy [2] | More variable estimates; struggles with templated changes [2] [3] | Cannot detect 'designer' mutations from HDR; miscalls alleles in clones [3] [7] |
| ICE | Acceptable accuracy [2] | More variable estimates [2] | Can miscall alleles in edited clones [7] |
| DECODR | Most accurate for most samples [2] | More accurate for identifying indel sequences [2] | --- |
| TIDER | --- | Specialized for knock-in efficiency of short epitope tags; outperforms others for this purpose [2] | Requires three sequencing traces (control, test, and a special reference) [3] |
| NGS (Gold Standard) | High accuracy and sensitivity [7] [4] | Comprehensive view of all editing outcomes [4] | Expensive, time-intensive, requires bioinformatics expertise [4] |
This comparative data underscores that while TIDE is a powerful tool, its results must be interpreted with caution, especially when analyzing complex editing outcomes. For validation of critical results, especially in clonal populations, confirmation with targeted next-generation sequencing (NGS) is recommended, as TIDE and other decomposition tools can miscall alleles in edited clones [7].
Experimental Protocols for Reliable TIDE Analysis
Adhering to standardized wet-lab and computational protocols is essential for generating high-quality data that TIDE can analyze effectively.
Sample Preparation and Sequencing
The following workflow details the critical steps for generating optimal sequencing traces.
Detailed Methodology:
- PCR Amplification: Isolate high-quality genomic DNA and amplify the target region using primers that span the edited site. It is crucial to use high-fidelity DNA polymerases with proofreading capabilities to minimize PCR errors, and to optimize conditions (e.g., annealing temperature) to ensure specific amplification of a single product [29]. Non-specific amplification is a major source of noisy, unreadable sequencing traces.
- Sanger Sequencing: Submit purified PCR products for capillary sequencing. As per TIDE recommendations, the target region for sequencing should be approximately 700 base pairs long, with the CRISPR cut site located preferably 200 base pairs downstream of the sequencing primer site [1]. This ensures a long, high-quality upstream sequence for accurate alignment between the control and test samples. Sequencing from both the forward and reverse strands is advisable to confirm any ambiguous results [29].
TIDE Analysis and Data Quality Control
Once sequencing is complete, the trace files are analyzed using the TIDE web tool.
Table 2: Essential Research Reagent Solutions for TIDE Analysis
| Reagent / Material | Function & Importance |
|---|---|
| High-Fidelity DNA Polymerase | Ensures accurate PCR amplification of the target locus, minimizing polymerase-induced errors that corrupt sequencing traces [29]. |
| Purified Genomic DNA Template | High-quality, intact DNA is foundational. Degraded or impure DNA templates lead to low-quality sequencing traces and erroneous base calls [29]. |
| Sanger Sequencing Service/Kit | Generates the raw chromatogram (trace) files. The quality of the sequencing run itself, including signal intensity and low background noise, is critical [1]. |
| Wild-Type Control DNA | Provides the reference sequence for the TIDE algorithm to decompose the edited sample trace. Must be from an unedited, isogenic source [9]. |
| TIDE Web Tool / Algorithm | The decomposition engine that quantifies indel frequencies and spectra. Requires specific input (e.g., .ab1 or .scf files, sgRNA sequence) [1]. |
TIDE Workflow and QC: The analysis involves uploading the control and test sample chromatogram files along with the sgRNA sequence to the TIDE web tool [1]. Before trusting the results, researchers must consult the "Quality control - Aberrant sequence signal" plot generated by TIDE. This plot visualizes the percentage of aberrant nucleotide signals along the sequence trace. A successful experiment will show a low, evenly distributed signal in the control sample and a clear increase in the aberrant signal downstream of the cut site in the test sample [1]. If the aberrant signal is high before the cut site in the test sample, the data quality is likely too poor for a reliable analysis.
A Scientist's Toolkit for Addressing Poor Data Quality
When faced with suboptimal TIDE results, a systematic troubleshooting approach is necessary.
Actionable Recommendations:
- If chromatograms show signal decay or high noise: Re-optimize your PCR conditions to ensure specificity and yield. Consider using a different primer set for sequencing.
- If the TIDE aberrant signal is high in the control sample or before the cut site in the test sample: The sequencing reaction itself may have failed, or the PCR product may be impure. Repeating the sequencing reaction or re-purifying the PCR product is advised [1] [29].
- If the decomposition fails (R² < 0.9) despite seemingly good traces: The editing may be too complex for TIDE's decomposition window. Consider using an alternative tool like DECODR, which was found to be more accurate for identifying complex indel sequences, or TIDER if analyzing knock-ins [2]. For definitive results, especially when characterizing clonal cell lines, targeted NGS remains the gold standard [7] [4].
In conclusion, securing high-quality Sanger sequencing traces is not merely a preliminary step but the very foundation of a successful TIDE analysis. By adhering to rigorous experimental protocols, systematically validating data quality through the tool's built-in controls, and understanding the comparative strengths and weaknesses of alternative methods, researchers can confidently employ TIDE to obtain accurate, publication-quality data on their genome editing experiments.
The Tracking of Indels by Decomposition (TIDE) method has established itself as a fundamental technique in the genome editing workflow, providing researchers with a rapid and cost-effective means to quantify the efficacy and spectrum of mutations induced by programmable nucleases such as CRISPR-Cas9 [9]. As a computational tool that deconvolutes Sanger sequencing trace data from edited cell populations, TIDE represents a significant advancement over earlier, less quantitative methods like the T7 endonuclease I (T7E1) assay, which suffers from limited dynamic range and inability to provide sequence-level information [7]. The accuracy and reliability of TIDE analysis, however, are profoundly influenced by two critical user-defined parameters: the indel size range and the P-value threshold [1].
Proper configuration of these parameters is not merely a technical formality but a prerequisite for obtaining biologically meaningful results. The indel size range determines the spectrum of mutations the algorithm will attempt to identify, while the P-value threshold sets the statistical stringency for accepting these mutations as genuine [1]. Within the broader context of TIDE assay research, understanding the interplay between these parameters and their optimization for specific experimental scenarios is essential for researchers, scientists, and drug development professionals who rely on accurate quantification of genome editing outcomes [2]. This guide provides a comprehensive comparison of TIDE performance relative to alternative tools, with specific emphasis on how parameter selection influences analytical outcomes across different editing scenarios.
TIDE Parameters: Theoretical Framework and Default Settings
Core Algorithmic Parameters and Their Functions
The TIDE algorithm operates by comparing Sanger sequencing chromatograms from control (unedited) and experimental (edited) samples, decomposing the complex mixture of sequences into constituent wild-type and indel-containing alleles [9]. Two parameters directly control the scope and statistical rigor of this decomposition process.
Indel Size Range: This parameter defines the maximum size of insertions and deletions that TIDE will model during its decomposition analysis [1]. The default value is typically set to 10 base pairs [1]. This range determines the window of possible mutations the algorithm considers when attempting to explain the deviations observed in the sequencing trace of the edited sample. Setting this value too low risks missing larger indels, while setting it unnecessarily high may introduce noise and reduce computational efficiency.
P-value Threshold: This parameter sets the significance cutoff for the decomposition results [1]. The default value is typically p < 0.001. This stringent threshold ensures that only indels with strong statistical support are included in the final output, minimizing false positives. The P-value is derived from statistical tests that evaluate how well each identified indel explains the observed sequence trace data compared to the wild-type reference.
Practical Implementation in the TIDE Web Tool
When using the TIDE web interface, these parameters are adjustable through the 'advanced settings' option [1]. The algorithm also utilizes additional fixed parameters that work in concert with the user-defined settings:
- Alignment Window: The sequence segment used to align control and test samples (default left boundary: 100 bp) [1]
- Decomposition Window: The sequence segment used for the decomposition analysis (default: maximum possible window) [1]
These parameters collectively ensure that the decomposition focuses on the relevant region surrounding the nuclease cut site, which is typically located between nucleotides 17 and 18 in the sgRNA sequence [1].
Comparative Performance of TIDE and Alternative Tools
Systematic Comparison of Computational Tools
Recent systematic evaluations have revealed important insights about the performance characteristics of TIDE and alternative indel analysis tools. A 2024 study compared four web tools—TIDE, ICE, DECODR, and SeqScreener—using artificial sequencing templates with predetermined indels, providing unprecedented quantitative assessment of their accuracy and limitations [2].
The study demonstrated that these tools generally estimate indel frequency with acceptable accuracy when the indels are simple and contain only a few base changes. However, the estimated values became more variable among the tools when the sequencing templates contained more complex indels or knock-in sequences [2]. A key finding was that although all tools effectively estimated net indel sizes, their capability to deconvolute specific indel sequences exhibited variability with certain limitations.
Table 1: Overall Performance Comparison of CRISPR Analysis Tools
| Tool | Best Application | Strengths | Key Limitations |
|---|---|---|---|
| TIDE | Non-templated Cas9 editing [3] | Rapid assessment; quantitative indel spectrum [9] | Limited detection of large indels (>50 bp) [5] |
| TIDER | Template-directed editing [30] [3] | Quantifies templated mutations plus non-templated indels [30] | Requires additional reference sequencing trace [3] |
| ICE | General indel analysis [4] | User-friendly interface; batch sample upload [4] | Comparable limitations for complex indels [2] |
| DECODR | Complex editing patterns [2] | More accurate for majority of samples; handles larger indels [2] [5] | Less established in community |
| PtWAVE | Large deletion analysis [5] | Detects indels up to 200 bp; superior accuracy for large deletions [5] | Newer tool with less extensive validation |
Parameter-Specific Performance Metrics
The performance of TIDE is particularly dependent on appropriate parameter setting for different experimental scenarios. The 2024 comparative study revealed that DECODR provided the most accurate estimations of indel frequencies for the majority of samples, consistent with previous reports [2]. Meanwhile, TIDE-based TIDER was found to outperform other tools for estimating knock-in efficiency of short epitope tag sequences [2].
Table 2: Parameter Optimization Guidelines for Different Editing Scenarios
| Editing Scenario | Recommended Tool | Optimal Indel Size Range | Optimal P-value | Reported Accuracy |
|---|---|---|---|---|
| Simple indels (1-3 bp) | TIDE or ICE | 10-15 bp (default adequate) [1] | p < 0.001 (default) [1] | Acceptable accuracy [2] |
| Complex indels | DECODR | 15-20+ bp | p < 0.001 | More accurate than TIDE [2] |
| Large deletions (>50 bp) | PtWAVE or DECODR | Up to 200 bp for PtWAVE [5] | p < 0.001 | Superior to TIDE [5] |
| Knock-in sequences | TIDER | 10-15 bp | p < 0.001 | Outperforms other tools [2] |
| Low-frequency editing | DECODR | 10-15 bp | p < 0.01 | More accurate in low range [2] |
For routine editing experiments with predominantly small indels, TIDE's default parameters (indel size range: 10; P-value: <0.001) generally yield acceptable results. However, when editing efficiency is extremely high or low, or when complex indels are anticipated, parameter adjustment and tool selection become critical factors for accurate quantification [2].
Experimental Protocols for Parameter Validation
Protocol for Assessing Optimal Indel Size Range
To empirically determine the optimal indel size range for a specific experimental system, researchers can employ the following validation protocol adapted from benchmark studies:
Sample Preparation: Generate edited cell pools using CRISPR-Cas9 with multiple sgRNAs targeting different genomic loci to create diverse indel spectra [2].
PCR Amplification: Amplify genomic regions surrounding the target sites using high-fidelity PCR master mixes (e.g., KOD One PCR Master Mix) with primers flanking the cut site [2].
Sequencing: Perform Sanger sequencing of PCR products using standard capillary sequencers [2] [9].
Parameter Testing: Analyze the same sequencing files with TIDE while systematically varying the indel size range parameter (e.g., 5, 10, 15, 20 bp).
Validation: Compare results with orthogonal methods such as targeted next-generation sequencing (considered the gold standard) or subcloning followed by Sanger sequencing [7].
Optimization: Select the indel size range that produces the highest concordance with validation methods while maintaining R² > 0.9 for decomposition goodness of fit [1].
This protocol was used in the 2024 comparative study, which employed artificial sequencing templates with predetermined indels to quantitatively assess tool performance [2].
Protocol for P-value Threshold Optimization
The P-value threshold can be optimized using a similar empirical approach:
Data Analysis: Process sequencing data from edited samples using a range of P-value thresholds (e.g., 0.01, 0.001, 0.0001).
False Positive Assessment: Include negative control samples (untransfected cells) to determine the false positive rate at each threshold.
Sensitivity Measurement: Use samples with known editing efficiencies (validated by NGS) to assess sensitivity across thresholds.
Threshold Selection: Select the P-value that minimizes false positives in controls while maintaining high sensitivity in edited samples.
Studies have indicated that the default P-value threshold of 0.001 generally provides a good balance between sensitivity and specificity [1], but this may vary depending on sequencing quality and editing complexity.
Research Reagent Solutions
Table 3: Essential Research Reagents for TIDE Analysis
| Reagent/Category | Specific Examples | Function in Workflow |
|---|---|---|
| CRISPR Components | Alt-R CRISPR-Cas9 crRNA (IDT) [2]; Alt-R S.p. Cas9 Nuclease V3 (IDT) [2] | Forms ribonucleoprotein complex for genome editing |
| PCR Amplification | KOD One PCR Master Mix (Toyobo) [2]; MyTaq Red Mix (Bioline) [9] | Amplifies genomic region surrounding target site |
| DNA Extraction | ISOLATE II Genomic DNA Kit (Bioline) [9] | Isolves high-quality genomic DNA for PCR |
| Sequencing | BigDye Terminator v3.1 (Applied Biosystems) [9] | Sanger sequencing reaction preparation |
| Positive Controls | Artificial sequencing templates [2] | Validates parameter settings and tool performance |
Workflow and Decision Pathway for Parameter Optimization
The following diagram illustrates the experimental workflow and decision process for optimizing TIDE parameters:
Diagram 1: Workflow for TIDE Parameter Optimization. This flowchart illustrates the experimental process and decision points for parameter adjustment, highlighting when validation and parameter refinement are necessary.
The optimization of indel size range and P-value threshold represents a critical step in ensuring accurate quantification of genome editing outcomes using TIDE analysis. While default parameters (indel size range: 10; P-value: <0.001) serve as a reasonable starting point for simple editing scenarios, complex indel patterns, large deletions, and knock-in experiments require careful parameter adjustment and, in some cases, alternative tools such as DECODR or TIDER [2].
The comparative data presented in this guide demonstrates that tool selection and parameter optimization must be guided by the specific experimental context and the nature of expected editing outcomes. As the field of genome editing continues to advance toward more complex applications, including therapeutic development, the precise quantification of editing outcomes through proper parameter optimization remains fundamental to research quality and reproducibility.
Researchers are encouraged to validate their parameter choices using orthogonal methods like next-generation sequencing, particularly when working with novel editing systems or experimental models where the indel spectrum may not be well characterized. This rigorous approach to parameter optimization ensures that TIDE analysis continues to provide valuable, quantitative insights into genome editing efficiency and specificity across diverse applications.
In the field of genome editing, accurately assessing the outcome of CRISPR-Cas9 experiments is crucial for both basic research and therapeutic development. The TIDE (Tracking of Indels by Decomposition) assay has emerged as a widely adopted method for quantifying the efficiency and spectrum of non-homologous end joining (NHEJ)-mediated indel mutations [8] [3]. This rapid, cost-effective technique utilizes Sanger sequencing data from edited and control samples, applying a decomposition algorithm to quantify the variety and frequency of induced insertions and deletions in a pooled cell population [9].
A critical quality metric within the TIDE analysis is the R² value, a statistical measure of goodness-of-fit that indicates how well the computational model explains the observed sequencing data [1]. A low R² value signals poor model fit, potentially leading to inconclusive or unreliable quantification of editing efficiency. This guide provides a systematic framework for researchers to diagnose, troubleshoot, and resolve the common issues that underlie low R² values in TIDE assays, and objectively compares its performance against other prevalent genome editing assessment methods.
Understanding the R² Value in TIDE Analysis
In the context of TIDE, the R² value represents the coefficient of determination. Simply put, it quantifies the proportion of the variance in the complex sequencing trace of the edited sample that is predictable from the combination of wild-type and indel sequence models identified by the algorithm [1] [31]. The TIDE software decomposes the mixed sequencing chromatogram into its constituent sequences (wild-type and various indels), and the R² value reflects how successfully this decomposition matches the actual data.
As a rule of thumb, an R² value greater than 0.9 is recommended for a reliable decomposition result [28] [1]. Values significantly below this threshold indicate that the model does not adequately account for the sequencing data, necessitating further investigation. The following diagram illustrates the key components of the TIDE workflow and the central role of the R² value as a quality checkpoint.
Systematic Troubleshooting of Low R² Values
A low R² value can stem from issues related to sample preparation, sequencing quality, or analysis parameters. The troubleshooting process should follow a logical progression, as outlined below.
Diagnostic Flowchart for Low R²
Step 1: Evaluate Sequencing Data Quality
The foundation of a successful TIDE analysis is high-quality Sanger sequencing data. The first step is to rigorously assess the quality of the sequencing chromatograms.
- Inspect the Aberrant Sequence Signal Plot: This plot, generated by the TIDE software, is the primary diagnostic tool. In a high-quality control sample (non-edited), the percentage of aberrant nucleotides should be low (ideally <10%) and evenly distributed along the entire sequence trace. A high or noisy aberrant signal in the control sample indicates underlying sequencing problems that will confound the analysis [1].
- Verify the Break Site Location: The plot should show a clear increase in the aberrant sequence signal (green line) around the expected Cas9 cut site (typically 3 bp upstream of the PAM sequence). If the signal increase does not align with the expected location, it may suggest off-target editing or problems with sample identification [1].
- Examine Chromatogram Quality: Visually inspect the raw sequencing chromatogram files for signs of poor quality, such as high background noise, overlapping peaks (peak compression), or rapid signal decay towards the end of the read [28] [1].
Remedial Actions:
- If the control sample shows high aberrant signal, the sequencing reaction must be repeated. Optimize the PCR purification step and use fresh sequencing reagents.
- Ensure the target region is sequenced to a length of ~700 bp, with the break site located approximately 200 bp downstream from the sequencing start site to provide sufficient high-quality sequence for robust alignment and decomposition [1].
Step 2: Optimize TIDE Analysis Parameters
If sequencing quality is confirmed, the next step is to adjust the analysis parameters within the TIDE web tool to better fit the specific data.
- Increase the Indel Size Range: The default setting models indels up to 10 bp in size. If your experiment has generated larger insertions or deletions, the algorithm will fail to account for them, resulting in a low R². Progressively increase the "Indel size range" parameter (e.g., to 15 or 20) to test if this improves the fit [28] [1].
- Adjust the Decomposition Window: The decomposition window is the sequence segment used for the computational analysis. The default is the largest possible window. However, if the ends of the sequencing read are of low quality or contain repetitive sequences, they can interfere with the analysis. Manually adjust the boundaries of the decomposition window to exclude low-quality regions while still capturing a representative sequence downstream of the break site [1].
- Review the Alignment Window: The alignment window (upstream of the break site) is used to align the control and test sequences. If the break site is too close to the start of the sequence read, you may need to reduce the left boundary from the default value of 100 to a lower number to find a high-quality alignment region [1].
Step 3: Address Biological and Experimental Factors
Certain biological and experimental factors can produce complex editing outcomes that are challenging for the TIDE algorithm to model perfectly.
- Highly Complex Indel Populations: The TIDE algorithm uses non-negative linear regression to resolve the mixture of sequences. If the edited cell pool contains an extremely high number of different indels, each at a low frequency, the model's ability to resolve them may be overwhelmed, leading to a lower R². This is a known limitation of the method compared to next-generation sequencing (NGS) [4].
- Unexpected Mutations: The presence of large deletions or complex structural variations that extend beyond the analyzed region will not be captured by TIDE, contributing to a poor fit [1] [4]. Furthermore, single-nucleotide polymorphisms (SNPs) in the target region can be misinterpreted as editing events, distorting the results.
- The 75% Aberrant Signal Limit: As discussed in researcher forums, even in a theoretical 100% edited population, the average aberrant sequence signal downstream of the cut site may only reach ~75%. This is because there is a 25% chance that a mutated sequence will, by chance, contain the same nucleotide as the wild-type sequence at any given position [28]. This inherent biological limitation means that perfect model fits are not always attainable.
Comparative Analysis of Genome Editing Assessment Methods
While TIDE is a powerful tool, understanding its performance relative to other methods is essential for selecting the right assay. The following table summarizes a comparative analysis of key techniques, with supporting quantitative data on their performance from a recent study [8].
Table 1: Comparative Analysis of CRISPR Genome Editing Assessment Methods
| Method | Principle | Throughput | Quantitative Nature | Indel Spectrum Info | Key Limitations |
|---|---|---|---|---|---|
| TIDE [8] [3] [1] | Decomposition of Sanger sequencing traces | Medium | Quantitative | Yes, identifies major indels | Accuracy drops with highly complex populations; cannot detect very large indels. |
| T7 Endonuclease I (T7EI) [8] [4] | Cleavage of heteroduplex DNA at mismatches | High | Semi-quantitative | No | Provides no information on the types of indels generated. |
| ICE [8] [4] | Advanced decomposition of Sanger traces | Medium | Quantitative | Yes, more comprehensive than TIDE | Proprietary algorithm (Synthego); performance dependent on sequencing quality. |
| ddPCR [8] | Fluorescent probe-based detection in partitioned droplets | High | Highly quantitative and precise | Limited, requires predefined probes | Requires specific probe design; cannot discover novel or unexpected indels. |
| Next-Generation Sequencing (NGS) [8] [4] | High-throughput sequencing of amplified target site | Low (cost and time per sample) | Highly quantitative and sensitive | Yes, provides the most comprehensive profile | Expensive, time-consuming, and requires advanced bioinformatics expertise. |
Table 2: Quantitative Performance Comparison from a Controlled Plasmid Model Study [8]
| Method | Reported Editing Efficiency at 50% Mixture | Correlation with NGS (R²) | Detection of Predominant Indel Types |
|---|---|---|---|
| TIDE | 48% | 0.92 | Yes |
| ICE | 51% | 0.96 | Yes |
| ddPCR | 50% | >0.99 | No |
| T7EI Assay | ~45% (semi-quantitative) | Not Reported | No |
The data from a controlled plasmid model study [8], which simulated different editing frequencies by mixing wild-type and edited plasmids in known ratios, demonstrates that TIDE provides a good approximation of true editing efficiency. However, its accuracy is slightly surpassed by ICE and significantly by ddPCR. It is important to note that in real-world genomic DNA samples, the presence of a more complex and unpredictable indel spectrum can widen this performance gap.
Detailed Experimental Protocol for TIDE Assay
A reliable TIDE result begins with a robust experimental workflow. The following protocol is standardized for human cells transfected with CRISPR-Cas9 components.
Step 1: Sample Preparation and Genomic DNA Extraction
- Cell Culture and Transfection: Culture the cells (e.g., K562, RPE) according to standard conditions. Transfect with the CRISPR-Cas9 plasmid(s) using an appropriate method (e.g., nucleofection for K562 cells, lipofection for adherent RPE cells) [9]. Include a control sample transfected without the nuclease or sgRNA.
- Genomic DNA Isolation: Harvest cells 2-4 days post-transfection. Isolate genomic DNA using a commercial kit (e.g., ISOLATE II Genomic DNA Kit, Bioline). Ensure DNA concentration and purity (A260/280 ratio of ~1.8) are measured using a spectrophotometer [9].
Step 2: PCR Amplification of the Target Locus
- Primer Design: Design primers that flank the CRISPR target site, generating a PCR product of ~700 bp. The Cas9 break site should be located ~200 bp downstream of the sequencing start site to ensure high-quality sequence data for the TIDE analysis [1] [9].
- PCR Reaction:
- PCR Product Purification: Purify the PCR product using a PCR clean-up kit (e.g., PCR ISOLATE II PCR and Gel Kit, Macherey-Nagel Gel and PCR Clean-Up Kit) to remove primers and enzymes [8] [9].
Step 3: Sanger Sequencing
- Sequencing Reaction: Use the same PCR primer (forward or reverse) for sequencing as was used for the initial PCR. A typical reaction uses ~30 ng of purified PCR product, 4 μL of BigDye Terminator v3.1, and 5 pmol of primer in a 20 μL reaction [9].
- Thermocycling for Sequencing:
- Initial Denaturation: 96°C for 1 minute.
- 30 cycles of: 96°C for 30s, 50°C for 15s, 60°C for 4 minutes [9].
- Purification and Run: Purify the sequencing reaction to remove unincorporated dyes and resuspend in Hi-Di formamide. Analyze the samples on a capillary sequencer (e.g., Applied Biosystems 3730xl DNA Analyzer) [9].
Step 4: TIDE Web Tool Analysis
- Data Input: Go to the TIDE web tool (http://apps.datacurators.nl/tide/). Input the 20nt sgRNA sequence (without the PAM). Upload the control and test sample sequencing files in
.ab1or.scfformat [1]. - Parameter Setting: Start with default parameters. If the R² value is low, systematically adjust the advanced settings as described in the troubleshooting section (e.g.,
Indel size range,Decomposition window). - Result Interpretation: Download the result report. A valid result should have an R² > 0.9. The report will detail the overall editing efficiency and a table of the specific indels identified with their frequencies and p-values [1].
Table 3: Key Research Reagent Solutions for TIDE Assay
| Item | Function / Description | Example Product / Vendor |
|---|---|---|
| High-Fidelity PCR Master Mix | Amplifies the target genomic locus with low error rate to prevent false positives. | Q5 Hot Start High-Fidelity Master Mix (NEB) [8] |
| Genomic DNA Isolation Kit | Purifies high-quality, intact genomic DNA from transfected cells. | ISOLATE II Genomic DNA Kit (Bioline) [9] |
| PCR Clean-up Kit | Purifies PCR products by removing excess primers, nucleotides, and enzymes before sequencing. | Gel and PCR Clean-Up Kit (Macherey-Nagel) [8] |
| BigDye Terminator Kit | Provides reagents for cycle sequencing, incorporating fluorescently labeled dideoxynucleotides. | BigDye Terminator v3.1 (Applied Biosystems) [9] |
| TIDE Web Tool | The online software that decomposes Sanger sequencing traces to quantify indel frequencies. | http://apps.datacurators.nl/tide/ [1] |
| Control Plasmid Mix | Validates the TIDE protocol and analysis by providing known ratios of edited/wild-type sequences. | Custom-designed plasmids (e.g., PLN R14del and WT reporters) [8] |
The TIDE assay remains a valuable method for the rapid and cost-effective quantification of CRISPR genome editing efficiency. Successfully navigating the challenge of low R² values requires a methodical approach that prioritizes high-quality input data. By rigorously checking sequencing chromatograms, strategically adjusting analysis parameters, and understanding the biological constraints of the system, researchers can confidently resolve inconclusive results.
For applications where extreme accuracy and comprehensive variant detection are paramount, such as in therapeutic drug development, orthogonal validation with a more sensitive method like NGS or ddPCR is strongly recommended. The choice of assay should be a deliberate one, balancing the need for speed and convenience against the requirements for sensitivity, precision, and depth of information.
The Tracking of Indels by Decomposition (TIDE) assay has emerged as a widely adopted method for rapid assessment of genome editing efficiency, particularly for quantifying small insertions and deletions (indels). However, a significant limitation of this technology is its inherent inability to detect large genetic modifications, including substantial deletions and megabase-scale chromosomal changes. This comprehensive analysis compares the performance of TIDE against emerging alternative methodologies, detailing their respective detection capabilities through experimental data and validation studies. We further provide detailed protocols for implementing these advanced detection strategies and visualize the critical workflow differences, equipping researchers with the necessary tools to overcome TIDE's primary constraint in comprehensive genome editing assessment.
The TIDE (Tracking of Indels by Decomposition) assay represents a breakthrough methodology that computationally deconvolutes Sanger sequencing data derived from bulk edited cell populations to estimate the efficiency and spectrum of targeted mutagenesis induced by programmable nucleases like CRISPR-Cas9, TALENs, and ZFNs [5]. By comparing sequencing chromatograms from edited samples against unedited controls, TIDE employs decomposition algorithms to quantify the relative abundance of different indel sequences without requiring clonal isolation or deep sequencing [1]. This approach provides researchers with a rapid, cost-effective solution for assessing editing efficiency, typically delivering results within 1-2 days compared to the several weeks often required for targeted deep sequencing [5].
Despite its widespread adoption and utility for basic editing assessment, TIDE contains fundamental limitations in its detection scope. The method's analytical window is inherently restricted to small indels, primarily those under 50 base pairs, with default parameters often set to detect indels of only up to 10 base pairs [1] [32]. This constrained detection range means that TIDE systematically fails to identify larger genetic alterations, including large deletions (LDs) exceeding 50-100 base pairs, large insertions, and more complex chromosomal rearrangements that frequently occur as unintended consequences of CRISPR-Cas9-induced double-strand breaks [5] [32]. The technology's limitation stems from its algorithmic design, which predefines a limited range of possible mutations during the deconvolution process, and expanding this range introduces significant uncertainty into the modeling [5]. Consequently, while TIDE remains valuable for initial efficiency estimates, its inability to detect these larger modifications poses substantial risks for data misinterpretation, particularly in therapeutic applications where comprehensive genotyping is critical for safety assessment.
Comparative Analysis of Detection Capabilities
Direct Comparison of Detection Ranges
The critical limitation of TIDE in detecting large gene modifications becomes evident when compared with specialized tools and methodologies designed specifically for this purpose. The following table summarizes the key differences in detection capabilities across available methods:
Table 1: Detection Capabilities of Genome Editing Assessment Methods
| Method | Maximum Deletion Detection | Primary Application | Key Limitations |
|---|---|---|---|
| TIDE | ~50 bp (limited) [5] | Small indel quantification in bulk populations | Cannot detect large deletions (>50 bp) or megabase-scale changes [1] [32] |
| PtWAVE | Up to 200 bp [5] | Wide-range indel detection including large deletions | Requires high-quality sequencing traces; newer method with less established track record |
| DECODR | Extended range beyond TIDE [5] | Improved detection of larger indels | Potential model uncertainty with range extension [5] |
| Long-Range PCR + Sequencing | Several kilobases [32] | Detection of large deletions and complex rearrangements | Low-throughput; requires specialized analysis |
| Targeted NGS | Limited by amplicon size (typically <500 bp) [32] | Comprehensive small indel profiling | Cannot detect very large deletions that exceed amplicon size [32] |
| Whole Genome Sequencing | Unlimited (theoretically) | Genome-wide detection of all modification types | Costly; computationally intensive; lower sensitivity for rare events |
As evidenced in Table 1, TIDE occupies the most restricted position in terms of deletion size detection, rendering it fundamentally unsuitable for comprehensive editing assessment where larger structural variations are of concern. This limitation becomes particularly problematic considering that large deletions exceeding 200 bp have been frequently reported at significant frequencies following CRISPR-Cas9 editing, with some studies indicating their occurrence can be substantial enough to impact experimental interpretations and therapeutic safety [32].
Experimental Validation of Limitations
Experimental evidence consistently demonstrates TIDE's failure to detect large on-target modifications. In one comprehensive evaluation, when CRISPR-Cas9 edited samples were analyzed using both TIDE and long-range sequencing methods, TIDE not only failed to detect large deletions but also produced inflated estimates of small indels due to its inability to account for these missing alleles in the decomposition algorithm [32]. This miscalculation occurs because large deletions eliminate the sequencing trace information downstream of the cut site that TIDE relies upon for its decomposition analysis, leading to systematic underestimation of total editing efficiency while simultaneously misrepresenting the mutation spectrum.
Quantitative comparisons reveal that TIDE's limitation is not merely theoretical but has practical consequences for editing assessment. In benchmarking studies using artificially mixed DNA samples containing known large deletions, TIDE demonstrated progressively diminishing accuracy for deletions exceeding 30 base pairs, with complete failure to detect deletions beyond 50 base pairs under standard parameters [5]. This performance deficit persists even when users manually extend the indel size range in TIDE's advanced settings, as the underlying algorithm becomes increasingly susceptible to signal noise and model uncertainty when more factors are considered [5]. Consequently, researchers relying exclusively on TIDE for editing assessment remain unaware of potentially significant large deletion events that could compromise experimental results or therapeutic applications.
Advanced Methodologies for Comprehensive Detection
PtWAVE: A High-Sensitivity Solution
The Progressive-type Wide-range Analysis of Varied Edits (PtWAVE) software represents a significant advancement specifically designed to overcome TIDE's limitations in large deletion detection [5]. This novel tool implements a progressively adjustable algorithm that systematically selects among various possible mutation patterns while evaluating mutation distributions using fitting algorithms with lower Bayesian information criterion (BIC), thereby constructing more reliable mutation distribution estimates even when detecting indels up to 200 base pairs [5]. Unlike TIDE, which employs fixed decomposition parameters, PtWAVE offers options for variable selection and fitting algorithms specifically engineered to prevent uncertainties in the model when extending the detection range.
Benchmarking studies directly comparing PtWAVE with TIDE and other decomposition tools demonstrate PtWAVE's superior performance for samples containing large deletions. When applied to in vitro capillary sequencing data mimicking DNA samples with large deletions, PtWAVE consistently achieved higher accuracy and sensitivity compared to existing TIDE analysis tools [5]. The software maintained stable analysis of trace sequencing data derived from actual genome-edited samples, confirming its practical utility for routine laboratory use. PtWAVE's enhanced detection capability stems from its implementation of a quality check based on sequence quality and distance, with specific algorithmic adjustments to determine optimal alignment windows that accommodate larger indels without compromising decomposition reliability [5].
Specialized Experimental Approaches
Beyond computational solutions, several experimental methodologies have been developed specifically to detect large on-target modifications that evade TIDE analysis:
Long-Range PCR and Sequencing: This approach utilizes PCR primers placed several kilobases apart flanking the target site, followed by sequencing of the resulting amplicons through either Sanger sequencing (for clonal analysis) or next-generation sequencing methods [32]. This enables direct detection of large deletions that would be missed by short-range PCR approaches. The method's primary limitation is its low-throughput nature and requirement for specialized analysis of the resulting data.
Digital PCR (ddPCR) and Quantitative Genotyping PCR (qgPCR): These methods employ specifically designed probe-based detection systems to identify and quantify predetermined large deletion events [32]. They offer high sensitivity and precise quantification but require prior knowledge of the specific deletion boundaries for probe design, making them unsuitable for discovery of novel large deletions.
Whole Genome Sequencing (WGS): As an unbiased approach, WGS theoretically enables detection of all deletion types regardless of size [32]. However, its practical implementation is limited by high cost, computational intensiveness, and lower sensitivity for detecting rare events in heterogeneous cell populations.
Single-Molecule Long-Read Sequencing: Technologies such as PacBio and Oxford Nanopore facilitate detection of large deletions by generating sequencing reads that can span entire deletion events, even those several kilobases in length [32]. These methods are particularly valuable for identifying complex rearrangements and precisely mapping deletion breakpoints.
Table 2: Methodologies for Detecting Large On-Target Modifications
| Methodology | Detection Principle | Size Range | Throughput | Key Applications |
|---|---|---|---|---|
| PtWAVE | Computational deconvolution of trace data | Up to 200 bp | High | Routine screening of large indels in bulk populations |
| Long-Range PCR + Sequencing | Amplification of large target regions | Several kilobases | Low-medium | Targeted validation of suspected large deletions |
| ddPCR/qgPCR | Probe-based quantification of specific events | Up to several kilobases | Medium | High-precision quantification of known large deletions |
| Whole Genome Sequencing | Unbiased genome-wide sequencing | Unlimited (theoretically) | Low | Comprehensive discovery of all variants |
| Long-Read Sequencing | Single-molecule sequencing of long DNA fragments | Up to tens of kilobases | Medium | Mapping complex structural variations |
Experimental Protocols for Comprehensive Assessment
PtWAVE Analysis Protocol
The PtWAVE protocol enables researchers to overcome TIDE's limitations through the following detailed workflow:
Input Data Preparation:
- Sequence both edited and unedited (WT) samples using standard capillary sequencing, saving trace files in .ab1 format.
- Prepare protospacer sequence (plain text) and PAM sequence (plain text) for CRISPR-Cas9 systems.
- Ensure sequencing quality meets standard thresholds (Phred score >30) for reliable decomposition [5].
Software Execution:
- Access the PtWAVE web interface at https://www.ptwave-ptbio.com/.
- Upload the sequencing trace files from both edited and unedited samples.
- Input the protospacer and PAM sequences to define the target locus.
- Select appropriate parameters for detection range (default detects up to 100 bp deletions, extendable to 200 bp).
- Choose fitting algorithms and variable selection methods based on data characteristics [5].
Results Interpretation:
- Review the comprehensive spectrum of indels detected, paying particular attention to larger deletion events (>50 bp).
- Assess the quality metrics provided by the software to ensure reliable decomposition.
- Export results for comparative analysis with other methods if necessary.
PtWAVE's algorithm specifically addresses TIDE's limitations by implementing a more sophisticated alignment window determination process that accounts for potential large indels. The starting point of the alignment window is determined as the 5' end base possessing a running mean of Phred Score higher than 30, while the endpoint becomes the position obtained by subtracting a margin value (calculated as the sum of 10 bp and the maximum anticipated indel size) from the position of the latest cut site [5]. This expanded window enables detection of larger events while maintaining analytical rigor.
Long-Range PCR and Sequencing Protocol
For direct detection of large deletions exceeding PtWAVE's range, implement the following protocol:
Primer Design:
- Design primers flanking the target site with positions placed 1-5 kilobases apart, depending on the expected deletion sizes.
- Verify primer specificity and ensure they do not bind within repetitive regions.
- Optimize primers for long-range PCR using high-fidelity polymerases.
PCR Amplification:
- Set up reactions using specialized long-range PCR kits with enhanced processivity.
- Use touchdown PCR cycling conditions to improve specificity for long amplicons.
- Include appropriate positive and negative controls.
Product Analysis:
- Resolve PCR products on agarose gels to identify size shifts indicative of large deletions.
- Purify products and subject to Sanger sequencing for small pools or clones.
- Alternatively, prepare libraries for next-generation sequencing to comprehensively characterize deletion spectra.
Data Interpretation:
- Map sequencing reads to reference genome to identify deletion boundaries.
- Quantify deletion frequencies based on read counts or band intensities.
- Correlate with functional outcomes where applicable.
This protocol directly addresses TIDE's blind spot by physically detecting large deletions through size-based separation, followed by molecular characterization. The approach is particularly valuable for validating suspected large deletion events initially suggested by indirect evidence such as TIDE's poor R² values or anomalous decomposition results [32].
Research Reagent Solutions
Table 3: Essential Research Reagents for Comprehensive Genome Editing Assessment
| Reagent/Software | Function | Application Context | Key Features |
|---|---|---|---|
| TIDE Software | Decomposition of Sanger sequencing traces | Rapid assessment of small indels (<50 bp) | Web-based; simple interface; fast results [1] |
| PtWAVE Software | High-sensitive deconvolution for large indels | Detection of indels up to 200 bp | Variable selection algorithms; BIC evaluation [5] |
| TIDER Software | Quantification of templated edits plus indels | HDR editing analysis | Requires additional reference sequencing trace [3] |
| Long-Range PCR Kits | Amplification of large target regions | Detection of large deletions (>1 kb) | High-fidelity enzymes with enhanced processivity |
| NGS Library Prep Kits | Preparation of sequencing libraries | Comprehensive variant discovery | Compatible with various input types and sizes |
| Digital PCR Systems | Absolute quantification of specific edits | Validation of predetermined large deletions | High precision; no standard curve needed [32] |
Workflow Visualization
Diagram 1: Comparative Workflow for Genome Editing Analysis. This workflow highlights the critical decision point between limited TIDE analysis and comprehensive approaches that detect large deletions.
The TIDE assay's inability to detect large deletions and megabase-scale changes represents a fundamental limitation that researchers must acknowledge when designing genome editing validation strategies. As demonstrated through comparative performance data and experimental validations, this constraint can lead to significant underestimation of total editing efficiency and mischaracterization of the mutation spectrum. Emerging solutions like PtWAVE and specialized experimental protocols provide viable pathways to overcome these limitations, enabling more comprehensive editing assessment. For researchers pursuing therapeutic applications, implementing these complementary methods is not merely optional but essential for accurate risk assessment and safety evaluation. The field continues to evolve toward integrated validation approaches that combine the speed and accessibility of decomposition tools like TIDE with the comprehensive detection capabilities of advanced methodologies, ultimately providing a more complete picture of genome editing outcomes.
Tracking of Indels by Decomposition (TIDE) has emerged as a widely adopted method for quantifying the efficiency and spectrum of insertions and deletions (indels) generated by genome editing tools such as CRISPR-Cas9, TALENs, and ZFNs [1] [9]. This computational approach deconvolves Sanger sequencing chromatograms from edited cell populations, providing researchers with a rapid, cost-effective alternative to more labor-intensive cloning sequencing or expensive next-generation sequencing (NGS) [33] [13]. The core innovation of TIDE lies in its decomposition algorithm, which mathematically resolves complex sequencing traces into their constituent indel sequences, quantifying both the identity and frequency of each mutation type present in the pooled sample [9].
The accuracy of TIDE analysis fundamentally depends on proper parameter configuration, particularly the setting of decomposition boundaries that define the genomic region analyzed for indel quantification [1]. These boundaries are not merely technical settings but represent critical methodological choices that determine which mutations are detected and how accurately they are quantified. While TIDE provides default parameters that work adequately for many standard applications, mastering the adjustment of decomposition boundaries enables researchers to extract more reliable and comprehensive mutation data from their genome editing experiments, particularly for challenging targets or when seeking to detect larger structural variations [33].
Theoretical Framework: The Role of Decomposition Boundaries
The TIDE algorithm operates through a sequential analytical process that begins with sequence alignment and culminates in the decomposition of mixed sequencing traces [1] [33]. Understanding this workflow is essential for making informed decisions about boundary adjustments.
Algorithmic Workflow of TIDE Analysis
The following diagram illustrates the key stages in the TIDE analysis process where boundary parameters exert their influence:
Functional Significance of Boundary Parameters
The decomposition window represents the sequence segment used for the core computational decomposition process [1]. This region is positioned downstream of the CRISPR-Cas9 cut site (typically located between nucleotides 17 and 18 of the sgRNA sequence) and must be carefully selected to balance detection sensitivity with analytical precision [1]. The default settings automatically configure the largest possible window based on the uploaded sequences, which generally provides the most robust estimation of mutations [1].
The alignment window functions as the sequence segment used to align control and test samples before decomposition analysis [1]. Proper alignment is crucial because it establishes the correct reading frame for subsequent indel detection and quantification. The default left boundary is set at 100 base pairs to avoid poor-quality base-calling at the start of Sanger sequences, while the right boundary automatically extends to the cut site minus 10bp [1].
The interdependency between these windows creates an analytical system where the alignment window ensures proper sequence registration, while the decomposition window enables the detection and quantification of mutations. When these boundaries are improperly set, the algorithm may either miss relevant mutations or incorporate noisy sequence data that reduces quantification accuracy.
Critical Scenarios for Boundary Adjustment
Addressing Suboptimal Sequencing Quality
Sequencing traces with localized quality degradation require strategic boundary adjustments to exclude problematic regions while retaining sufficient sequence for reliable decomposition. The following scenarios necessitate manual intervention:
- Low-quality sequence starts: When the beginning of the sequencing read shows consistently poor quality (Phred scores <30) extending beyond the default 100bp left boundary, the alignment window left boundary should be moved further downstream to the first region with stable, high-quality sequencing data [1] [33].
- Localized quality drops within decomposition window: If specific regions within the default decomposition window show severe quality deterioration (evidenced by high aberrant sequence signals in control samples), constricting the decomposition boundaries to exclude these regions improves analytical accuracy [1].
- Premature sequence termination: For shorter sequencing reads that end close to the cut site, both windows may need adjustment to ensure sufficient sequence context remains for proper analysis [1].
Expanding Mutation Detection Range
The standard TIDE configuration typically detects indels up to approximately 50 base pairs [33] [5]. However, CRISPR-Cas9 editing can generate larger deletions, particularly in certain cell types or genomic contexts [33]. To detect these larger structural variations:
- Extending decomposition window right boundary: Increasing the decomposition window size provides the necessary sequence context for detecting larger deletions [33].
- Adjusting indel size parameter: The indel size range parameter must be increased concurrently with decomposition window expansion to model larger mutations [1].
- Anticipating analytical trade-offs: While expanding detection range enables identification of larger indels, it may increase model uncertainty due to the incorporation of more variable sequence patterns [33].
Targeting Specific Genomic Contexts
Complex genomic regions with repetitive elements, homopolymer runs, or secondary structures often require customized boundary settings:
- Repetitive sequences: When repetitive elements flank the cut site, narrowing the decomposition window to exclude highly repetitive regions prevents misalignment and decomposition artifacts [1].
- Homopolymer regions: Sequencing traces through homopolymer tracts often show compressed peaks and elevated background signals, which can be excluded by boundary adjustment to improve quantification accuracy.
- High-GC regions: Areas with extreme GC content may exhibit sequencing artifacts that benefit from boundary refinement to maximize signal-to-noise ratio in the decomposition.
Experimental Protocols for Boundary Optimization
Systematic Approach to Boundary Configuration
Establishing optimal decomposition boundaries requires a methodical experimental approach:
Initial Quality Assessment
- Generate aberrant sequence signal plots for both control and test samples using default TIDE parameters [1].
- Identify regions where the control sample shows elevated aberrant signals (>10%), indicating inherent sequencing quality issues [1].
- Locate the area where test sample aberrant signals consistently diverge from the control, indicating the mutation start region [1].
Iterative Boundary Refinement
- Begin with the default decomposition window settings and document the resulting R² value and indel spectrum [1].
- Systematically adjust the left and right boundaries in 20-30bp increments, recording how each change affects the R² value and detected indel frequencies.
- Select the window configuration that maximizes R² value while maintaining a biologically plausible indel spectrum [1].
Validation Against Complementary Methods
Quantitative Assessment of Editing Efficiency Across Methods
The table below summarizes performance comparisons between TIDE and other commonly used genome editing assessment methods:
Table 1: Comparative Performance of Genome Editing Assessment Methods
| Method | Detection Range | Quantitative Accuracy | Time Required | Cost Considerations | Key Limitations |
|---|---|---|---|---|---|
| TIDE | Typically ≤50 bp indels [33] | High correlation with NGS for common indels (R²>0.9) [1] | 1-2 days [33] | Low (Sanger sequencing costs) | Limited for large deletions >50bp [33] |
| T7E1 Assay | Detects heteroduplex formation | Semi-quantitative, underestimates efficiency [8] [7] | 1 day | Very low | No sequence information, low dynamic range [7] |
| ICE | Similar to TIDE, up to ~30bp for single guide [33] | High correlation with NGS (R²=0.96) [4] | 1-2 days | Low (Sanger sequencing costs) | Limited for large indels [33] |
| ddPCR | Specific predefined edits | High precision for known mutations [8] | 1 day | Medium | Requires specific probe design, detects only targeted edits [8] |
| NGS | Comprehensive detection | Gold standard, detects all mutation types [8] [4] | Several weeks | High | Expensive, requires bioinformatics expertise [4] |
Advanced Applications and Specialized Scenarios
For researchers investigating specific biological questions or working with challenging editing systems, additional boundary considerations apply:
Multiplexed Editing Analysis
- When analyzing pools of cells edited with multiple sgRNAs, the decomposition window may need expansion to cover all potential cut sites.
- Larger indel size ranges should be specified to accommodate potential compound mutations between adjacent target sites.
Template-Directed Editing (TIDER)
- For homology-directed repair experiments, the related TIDER method requires additional sequencing traces and different boundary considerations to distinguish templated mutations from background indels [3] [13].
- The decomposition window must encompass both the Cas9 cut site and the incorporated templated sequence.
Large Deletion Detection with Next-Generation Tools
- Newer algorithms like PtWAVE extend detection capabilities to indels up to 200bp through improved decomposition approaches [33] [5].
- When using these advanced tools, decomposition windows must be sized accordingly, with careful attention to potential signal noise from expanded detection ranges [33].
Research Reagent Solutions for TIDE Analysis
Table 2: Essential Reagents and Materials for TIDE Workflow Implementation
| Reagent/Resource | Specification | Function in Workflow |
|---|---|---|
| Genomic DNA Isolation Kit | High-quality DNA from ≥1000 cells (e.g., ISOLATE II Genomic DNA Kit) [13] | Ensures comprehensive sampling of mutation diversity in edited cell pool |
| PCR Master Mix | High-fidelity polymerase (e.g., Q5 Hot Start High-Fidelity) [8] | Amplification of target region with minimal PCR bias |
| Sanger Sequencing Reagents | BigDye terminator kits [9] [13] | Generation of high-quality sequence chromatograms for decomposition |
| TIDE Web Tool | Accessible at https://tide.nki.nl or https://apps.datacurators.nl/tide/ [1] [3] | Core decomposition algorithm with adjustable boundary parameters |
| Control DNA Sample | Unedited wild-type sequence from identical genetic background [1] | Essential reference for proper sequence alignment and background subtraction |
Interpretation and Quality Control Metrics
Assessing Decomposition Quality
After boundary adjustment, researchers must evaluate the reliability of the decomposition results using multiple quality metrics:
- R² value: The goodness-of-fit statistic should exceed 0.9 for reliable decomposition [1]. Values below this threshold indicate poor model fitting, often due to suboptimal boundary settings or poor sequence quality.
- Aberrant sequence signal: Control samples should show low (<10%) and evenly distributed aberrant signals [1]. Test samples should display low signal before the break site and elevated signal downstream of the cut site [1].
- Indel spectrum plausibility: The reported indel frequencies should form a coherent biological pattern, typically centered around the cut site with decreasing frequency for larger indels.
Troubleshooting Suboptimal Decomposition
When decomposition quality remains poor despite boundary adjustment:
- Verify sequence quality: Ensure both control and test sequences have high-quality base calling throughout the regions used for alignment and decomposition.
- Check cut site positioning: Confirm the sgRNA sequence is correctly specified and the cut site is appropriately identified relative to the PAM sequence.
- Consider alternative algorithms: For persistent issues with large indel detection, consider transitioning to next-generation tools like PtWAVE or DECODR that offer expanded detection capabilities [33] [5].
Mastering the adjustment of decomposition boundaries represents an essential skill for researchers utilizing TIDE analysis in genome editing applications. The strategic configuration of these parameters enables optimization for diverse experimental scenarios, from standard indel quantification to detection of larger structural variations. As the field advances, next-generation decomposition tools like PtWAVE are overcoming traditional size limitations of TIDE, expanding detectable indel ranges to 200bp through improved algorithmic approaches [33] [5]. These developments, coupled with a deeper understanding of boundary optimization strategies, will continue to enhance the precision and utility of sequencing trace deconvolution for the comprehensive assessment of genome editing outcomes.
TIDE vs. Other Methods: A Critical Comparison of CRISPR Analysis Tools
In the realm of genome editing, successful manipulation of DNA sequences using tools like CRISPR-Cas9, TALENs, and ZFNs depends not only on efficient delivery of editing components but also on accurate assessment of the resulting modifications. The choice of validation method significantly impacts the reliability, efficiency, and cost-effectiveness of research and therapeutic development. The TIDE (Tracking of Indels by Decomposition) assay and T7 Endonuclease I (T7E1) mismatch detection assay represent two fundamentally different approaches to evaluating editing efficiency. Where TIDE offers sophisticated computational analysis of sequencing data for precise quantification, T7E1 provides a rapid, gel-based detection system for initial screening. This guide provides an objective comparison of these methodologies, supported by experimental data, to inform researchers and drug development professionals in selecting the optimal validation strategy for their specific applications within the broader context of indel analysis research.
T7 Endonuclease I (T7E1) Assay: A Semi-Quantitative Mismatch Detection Method
The T7E1 assay is an enzyme-based method that detects the presence of insertions or deletions (indels) resulting from non-homologous end joining (NHEJ) repair of CRISPR-induced double-strand breaks. The assay operates on the principle that the T7 Endonuclease I enzyme recognizes and cleaves heteroduplex DNA formed by hybridization between wild-type and indel-containing sequences.
Experimental Protocol for T7E1 Assay:
- PCR Amplification: The genomic target region encompassing the CRISPR cut site is amplified by PCR from both edited and control samples [8].
- Heteroduplex Formation: The PCR products are denatured by heating and then slowly re-annealed by cooling. This process allows for the formation of heteroduplexed DNA when indel-containing strands pair with wild-type strands, creating bulges at mismatch sites [7].
- Enzymatic Digestion: The re-annealed DNA is incubated with T7 Endonuclease I, which cleaves the heteroduplexes at the mismatch sites [8] [7].
- Visualization and Analysis: The digestion products are separated by agarose gel electrophoresis. Cleaved fragments are visualized and compared to the undigested parental band. Editing efficiency is estimated through densitometric analysis of band intensities using the formula: % gene modification = (1 - (1 - (sum of cleaved band intensities / sum of all band intensities))^(1/2)) × 100 [7].
TIDE (Tracking of Indels by Decomposition): A Quantitative Computational Approach
TIDE is a sequencing-based method that provides a detailed profile of indel spectra and frequencies by computationally decomposing Sanger sequencing chromatograms from edited cell pools. The algorithm compares sequence traces from edited and control samples to detect shifts in the sequencing profile caused by insertions or deletions.
Experimental Protocol for TIDE:
- PCR and Sequencing: The target site is amplified by PCR from both control (non-edited) and experimentally edited samples. These PCR products are then subjected to standard Sanger sequencing, generating chromatogram files (e.g., in .ab1 or .scf format) [1] [25].
- Data Upload and Parameter Configuration: The sequencing files from both samples, along with the sgRNA target sequence (excluding the PAM site), are uploaded to the TIDE web tool. Key parameters include specifying the cut site (typically 3 bases upstream of the PAM) and setting the decomposition window [1].
- Computational Analysis: The TIDE algorithm performs two critical operations [1]:
- Sequence Alignment: It aligns the control and test sample sequences using a predefined alignment window (typically starting ~100 bp from the sequence start to avoid poor-quality base-calling regions).
- Trace Decomposition: It decomposes the mixed sequence traces from the edited sample in the decomposition window (downstream of the cut site) into their constituent wild-type and indel-containing sequences using non-negative linear modeling.
- Output and Interpretation: The tool generates several outputs, including:
- Indel Spectrum Plot: Depicts the types and frequencies of identified indels [1].
- Aberrant Sequence Signal Plot: Shows the percentage of non-expected nucleotides along the sequence, with a noticeable increase after the cut site in successfully edited samples [1].
- Quality Metrics: An R² value indicates the goodness of fit of the decomposition model, with values >0.9 considered reliable [1].
Head-to-Head Comparison: Performance, Data, and Applications
Direct Performance Benchmarking and Experimental Validation
Multiple independent studies have systematically compared the accuracy and reliability of TIDE and T7E1 against reference methods like next-generation sequencing (NGS). The collective evidence demonstrates clear differences in their quantitative capabilities.
| Method | Quantitative Capability | Sensitivity to Low Indels | Accuracy at High Efficiency | Dynamic Range |
|---|---|---|---|---|
| T7E1 Assay | Semi-quantitative, underestimates efficiency [7] | Poor (often misses <10% indels) [7] | Poor (underestimates >30% efficiency) [7] | Narrow, non-linear |
| TIDE Assay | Fully quantitative, high correlation with NGS [10] [4] | Good for mid-range frequencies [10] | Good, comparable to NGS for pools [7] | Wide, linear |
A landmark study directly compared T7E1 and NGS at 19 genomic loci in human and mouse cells, revealing significant discrepancies. The T7E1 assay reported an average editing efficiency of 22%, while NGS revealed the actual average efficiency was 68% [7]. The T7E1 assay failed to detect activity for sgRNAs with less than 10% editing by NGS and substantially underestimated efficiency for highly active sgRNAs (>90% by NGS), reporting them as only modestly active [7]. Furthermore, sgRNAs that appeared to have similar activity by T7E1 (~28%) showed dramatically different actual efficiencies by NGS (40% vs. 92%) [7]. This confirms that T7E1 signals correlate more strongly with indel complexity than with true frequency, leading to unreliable quantification, especially with a single dominant indel [10].
In contrast, TIDE demonstrates significantly better performance. In evaluations, TIDE and similar computational tools showed much higher agreement with NGS data for estimating editing efficiencies in cell pools [7] [10]. While one study noted that TIDE could miscall alleles in edited clones, its performance for analyzing heterogeneous pools of cells is robust [7]. The decomposition algorithm successfully reconstructs the spectrum of indels from sequence traces, providing both frequency and identity of the predominant mutations [1] [25].
Information Output and Technical Specifications
The nature and depth of information provided by each method vary considerably, impacting their utility for different research objectives.
| Feature | T7E1 Assay | TIDE Assay |
|---|---|---|
| Indel Quantification | Estimated frequency only | Precise frequency for each indel type |
| Sequence Information | No sequence-level data | Identifies predominant indel sequences |
| Indel Size Detection | Limited resolution | Default up to 10 bp (adjustable) |
| Multiplexing | Not applicable | Batch analysis available |
| Primary Output | Gel image with bands | Indel spectrum plot, aberrant sequence signal plot, statistical summary |
| Key Limitation | Cannot differentiate between different types of indels | Less effective for very complex indel mixtures or knock-in analysis [10] |
The T7E1 assay fundamentally cannot provide nucleotide-level information about the induced mutations. It only indicates that a mutation is present based on heteroduplex formation and cleavage [4]. Conversely, TIDE specifically identifies the sequences of the most prevalent indels in the pool and their individual frequencies, offering invaluable insight for applications where the specific genetic outcome matters [1].
Practical Considerations: Workflow and Resource Requirements
Practical implementation factors often influence method selection as much as technical performance.
| Consideration | T7E1 Assay | TIDE Assay |
|---|---|---|
| Hands-On Time | Moderate (PCR, digestion, gel run) | Moderate (PCR, sequencing prep) |
| Total Turnaround Time | 1-2 days | 2-3 days (includes sequencing) |
| Cost per Sample | Low [25] | Moderate (cost of Sanger sequencing) [25] |
| Equipment Needs | Standard molecular biology lab (thermocycler, gel rig) | Access to Sanger sequencing facility |
| Technical Expertise | Basic molecular biology skills | Basic computational literacy for web tool |
| Throughput | Low to moderate | Moderate to high (especially with batch TIDE) |
| Best Suited For | Quick confirmation of nuclease activity, low-budget initial screening [4] | Precise optimization of editing conditions, quantitative assessment of efficiency |
The T7E1 assay is notably cost-effective and rapid, requiring only standard laboratory equipment [25]. TIDE, while more expensive than T7E1, remains significantly cheaper than clonal sequencing (~25-50 fold) or NGS (~100-fold for non-multiplexed runs), offering an excellent balance between cost and information depth [25].
Successful implementation of either assay requires specific reagents and computational resources. The following table details the key solutions and their functions.
| Reagent/Resource | Function in Assay | Specific Example/Note |
|---|---|---|
| T7 Endonuclease I | Cleaves heteroduplex DNA at mismatch sites | Commercial enzymes (e.g., NEB M0302) [8] |
| High-Fidelity PCR Master Mix | Amplifies the target genomic locus with high accuracy | e.g., Q5 Hot Start High-Fidelity Master Mix [8] |
| Agarose Gel Electrophoresis System | Separates and visualizes digested and undigested PCR products | Standard lab setup with ethidium bromide or GelRed [8] |
| Sanger Sequencing Service | Generates sequence chromatograms for decomposition analysis | Core facility or commercial service (.ab1 or .scf format) [1] |
| TIDE Web Tool | Decomposes sequencing traces to quantify indel spectrum and frequency | Free online resource (apps.datacurators.nl/tide) [1] |
| sgRNA Sequence | Essential parameter for TIDE analysis to define the expected cut site | 20nt sequence immediately upstream of PAM, without the PAM itself [1] |
The comparative analysis unequivocally demonstrates that TIDE and T7E1 serve distinct purposes in the genome editing workflow. The choice between them should be strategically aligned with the experimental goals, required data quality, and available resources.
Choose the T7E1 Assay if: Your primary need is a fast, low-cost, binary answer to the question "Did editing occur?" during initial gRNA screening or protocol optimization. It is ideal for quick checks where precise quantification and the nature of the indels are not critical [4].
Choose the TIDE Assay if: Your research requires accurate quantification of editing efficiency and insight into the spectrum of mutations generated. It is the superior tool for optimizing editing conditions, comparing nuclease performance, and for any application where knowing the specific indels and their frequencies is important for downstream analysis or validation [1] [25].
For the most demanding applications where comprehensive detection of all edit types—including complex knock-ins or very heterogeneous indels—is required, targeted next-generation sequencing remains the gold standard, albeit at a higher cost and complexity [8] [4]. However, for the vast majority of routine genome editing validation tasks, TIDE provides an optimal balance of quantitative precision, cost-effectiveness, and detailed output, solidifying its role as an indispensable tool in modern molecular biology and therapeutic development.
The advent of CRISPR-Cas9 technology has revolutionized modern biological research, making genome editing more accessible and efficient than ever before. However, successful genome editing depends critically on the ability to accurately assess the efficiency of programmable nucleases, most commonly by estimating the occurrence of insertions and deletions (indels) caused by nuclease-induced double-strand breaks. Among the various methods developed for this purpose, computational tools that analyze Sanger sequencing trace data have gained significant popularity due to their user-friendly nature and cost-effectiveness compared to next-generation sequencing (NGS). Two of the most prominent tools in this category are TIDE (Tracking of Indels by Decomposition) and ICE (Inference of CRISPR Edits). This review provides a comprehensive comparative analysis of these two platforms, examining their methodologies, performance characteristics, and suitability for different experimental contexts within the broader framework of TIDE assay research. As the field progresses, understanding the strengths and limitations of each tool becomes paramount for researchers, particularly those in drug development who require robust and reliable validation of gene editing outcomes.
At a Glance: Comparative Features of TIDE and ICE
The following table summarizes the core characteristics, capabilities, and requirements of TIDE and ICE based on current literature and developer specifications.
| Feature | TIDE (Tracking of Indels by Decomposition) | ICE (Inference of CRISPR Edits) |
|---|---|---|
| Primary Function | Quantifies spectrum and frequency of small indels from Sanger sequencing data [1] [3] | Determines editing efficiency, types, and relative abundances of indels [34] |
| Input Requirements | Two Sanger sequencing traces (control and edited sample) [1] | Sanger sequencing files for edited and control samples [34] |
| Key Output Metrics | Indel frequency, indel spectrum, R² value [1] | Indel percentage, Knockout Score, Knock-in Score, Model Fit (R²) [34] |
| Supported Nuclease Systems | CRISPR-Cas9, TALENs, ZFNs [1] | SpCas9, hfCas12Max, Cas12a, MAD7 [34] |
| Theoretical Basis | Sequence trace decomposition via non-negative linear regression [1] [6] | Algorithm using lasso regression [6] |
| Handling of Complex Edits | Limited with complex indels or knock-ins; TIDER needed for templated edits [2] [3] | Better capability for complex edits from multiple gRNAs or various nucleases [34] |
| Batch Analysis | Available through a separate batch site [1] | Native support for batch analysis of hundreds of samples [34] |
| Reported Accuracy | Accurate for simple indels; variable for complex indels [2] | Highly comparable to NGS (R² = 0.96) [34] [4] |
Performance and Accuracy in Experimental Settings
Insights from Systematic Comparisons
Recent systematic studies have shed light on the performance characteristics of both TIDE and ICE under controlled conditions. A 2024 comparative study that utilized artificial sequencing templates with predetermined indels demonstrated that both tools could estimate indel frequency with reasonable accuracy when the indels were simple and contained only a few base changes. However, the estimated values became more variable between the tools when the sequencing templates contained more complex indels. Among the tools tested, DECODR provided the most accurate estimations for the majority of samples, but ICE generally performed robustly [2]. Another independent study analyzing somatic CRISPR/Cas9 tumor models reported high variability in the number, size, and frequency of indels across different software platforms, including TIDE and ICE. This suggests that the specific experimental context, particularly the presence of larger indels common in in vivo models, can significantly impact tool performance [6].
A critical real-world case study comparing Oxford Nanopore sequencing to Sanger-based methods found that the indel frequencies from nanopore sequencing (analyzed with nCRISPResso2) closely mirrored those from both TIDE and ICE. Notably, the authors observed that "nCRISPResso2 exhibited closer alignment with ICE results than with TIDE, or even between TIDE and ICE themselves" [35]. This indicates that while both tools are useful, they can yield divergent results from the same sample.
Comparison to the Gold Standard
When validated against the gold standard of next-generation sequencing (NGS), ICE has demonstrated a strong correlation. Synthego reports that ICE analysis results are highly comparable to NGS, with an R² value of 0.96 [34] [4]. This high degree of concordance provides users with confidence in the quantitative output of the ICE tool. While TIDE also provides quantitative data, its accuracy can be more variable, particularly for samples with indel frequencies at the low or high extremes of the spectrum or with more complex editing patterns [2] [7].
Comparative Workflow and Validation. This diagram illustrates the shared experimental workflow for TIDE and ICE analysis, culminating in validation against Next-Generation Sequencing (NGS), the gold standard. ICE shows a reported high correlation (R² = 0.96) with NGS results [34] [4].
Experimental Protocols and Methodologies
Sample Preparation and Sequencing
The foundational steps for both TIDE and ICE analysis are identical, requiring high-quality starting material and precise amplification of the target locus.
- CRISPR Delivery and DNA Extraction: Introduce CRISPR components (e.g., Cas9 nuclease and guide RNA) into your target cells using your preferred method (e.g., nucleofection, lipofection). After a suitable incubation period (typically 3-4 days), harvest cells and extract genomic DNA using a standard protocol [2] [7].
- PCR Amplification: Design primers flanking the CRISPR target site. The amplicon should be large enough to provide a good sequencing read but typically around 700 bp is advised, ensuring the cut site is preferably located ~200 bp downstream from the sequencing start site [1]. Perform PCR amplification using a high-fidelity DNA polymerase to minimize errors.
- Sanger Sequencing: Purify the PCR products and submit them for Sanger sequencing using one of the PCR primers. The output must be the raw chromatogram file in
.ab1or.scfformat for both the edited sample and an unedited control sample [1] [34].
Computational Analysis Workflow
| Step | TIDE Protocol | ICE Protocol |
|---|---|---|
| Data Input | Upload control and test .ab1 or .scf files. Enter the sgRNA sequence (20nt, excluding PAM). TIDE assumes a cut between nucleotides 17 and 18 [1]. |
Upload control and edited .ab1 files. Enter the gRNA target sequence and select the nuclease used from a dropdown menu [34]. |
| Parameter Configuration | Adjustable parameters include the alignment window, decomposition window, indel size range, and p-value threshold. Default settings are often sufficient [1]. | Designed to be user-friendly with no parameters requiring optimization. The tool automatically processes the data once inputs are provided [34] [4]. |
| Interpretation of Results | Evaluate the Indel spectrum plot and R² value (aim for >0.9). Check the "Aberrant sequence signal" plot for quality control [1]. | Review the summary table for Indel %, KO Score, and R². Use detailed tabs (Traces, Alignment) for in-depth analysis of indel contributions [34]. |
Successful indel analysis requires not only software but also specific laboratory reagents and resources. The following table details key solutions and their functions in the experimental workflow.
| Research Reagent Solution | Function in CRISPR Validation |
|---|---|
| Programmable Nuclease (e.g., Cas9 Protein) | Creates a double-strand break at the specific genomic target site guided by the gRNA [2]. |
| Guide RNA (gRNA/crRNA) | Directs the Cas nuclease to the target DNA sequence via complementarity [2] [34]. |
| High-Fidelity DNA Polymerase | Accurately amplifies the target genomic region from extracted DNA with minimal PCR errors [2] [6]. |
| Sanger Sequencing Service | Generates the chromatogram sequence traces of the PCR amplicons, which serve as the primary data for TIDE/ICE analysis [1] [34]. |
| Wild-Type Control DNA | Provides a reference sequence from unedited cells, which is crucial for the decomposition algorithm to identify mutations [2] [1]. |
Tool Selection Logic. This decision diagram outlines key experimental factors to guide researchers in choosing between TIDE and ICE, including project goal, nuclease type, and throughput needs [2] [34] [3].
The choice between TIDE and ICE is not a matter of one being universally superior, but rather of selecting the right tool for the specific experimental context. For straightforward knockout experiments using SpCas9, where cost-effectiveness and simplicity are priorities, TIDE remains a reliable and established choice. Its strength lies in its focused application for quantifying small indels. However, for more complex editing scenarios—including those involving multiple gRNAs, alternative nucleases like Cas12a, or high-throughput screening workflows—ICE presents a more robust and versatile solution. Its higher correlation with NGS data, user-friendly interface requiring minimal parameter adjustment, and built-in batch processing capabilities make it particularly suited for modern, scalable genome editing projects, especially in drug development pipelines where reproducibility and accuracy are paramount.
Future developments in this field will likely focus on increasing the accuracy and scope of these computational tools, particularly for characterizing complex edits and knock-ins. Furthermore, as long-read sequencing technologies like Oxford Nanopore continue to decrease in cost, they may offer a compelling alternative by providing comprehensive indel characterization from a single run, potentially surpassing the limitations of Sanger-based decomposition methods [35]. Regardless of the platform, the fundamental requirement for careful experimental design, including proper controls and high-quality sequencing data, will remain the cornerstone of valid CRISPR analysis.
The advent of CRISPR-Cas9 genome editing has revolutionized biological research and therapeutic development, creating a critical need for robust methods to analyze editing outcomes. Two primary techniques have emerged for assessing CRISPR efficiency and characterizing the resulting insertion and deletion mutations (indels): the TIDE (Tracking of Indels by Decomposition) assay and Next-Generation Sequencing (NGS). While TIDE offers a rapid, cost-effective approach by computationally decomposing Sanger sequencing chromatograms, NGS provides a comprehensive, high-resolution view through massively parallel sequencing. This guide objectively compares the performance, applications, and practical considerations of these methods to help researchers and drug development professionals select the appropriate tool for their specific genome editing projects.
Methodological Principles and Workflows
TIDE Assay Workflow and Principles
The TIDE method simplifies the quantification of genome editing efficiency through a streamlined, PCR-based approach followed by specialized computational analysis. The protocol involves:
- PCR Amplification: The genomic region flanking the CRISPR target site is amplified from both edited and control (unmodified) cell populations [9] [25].
- Sanger Sequencing: The purified PCR products are subjected to standard capillary (Sanger) sequencing [9] [25].
- Computational Decomposition: The resulting sequencing chromatogram files (in .ab1 or .scf format) from the edited sample and the wild-type control are uploaded to the TIDE web tool. The algorithm aligns the sgRNA sequence to the control sequence to pinpoint the Cas9 cut site, typically located 3 base pairs upstream of the Protospacer Adjacent Motif (PAM). It then performs a decomposition analysis, comparing the edited sample sequence trace to the reference trace to identify the spectrum and relative abundance of different indels. The output includes the overall editing efficiency and a detailed breakdown of individual mutation types and their frequencies [9] [8].
The following diagram illustrates the key steps in the TIDE workflow and its underlying analytical principle:
Diagram 1: TIDE workflow and analytical principle.
Next-Generation Sequencing Workflow and Principles
NGS represents a fundamentally different approach, characterized by its massively parallel sequencing capability. The standard workflow for targeted NGS in CRISPR analysis includes:
- Library Preparation: DNA is fragmented, and platform-specific adapter sequences are ligated to the ends of the fragments. These adapters enable the DNA to bind to the sequencing flow cell and serve as priming sites for amplification and sequencing [36].
- Cluster Generation (Bridge PCR): The library is loaded onto a flow cell, where individual DNA fragments bind to the surface and are amplified in situ into clonal clusters through bridge amplification. This step creates millions of distinct DNA colonies, each representing a single original molecule, which is necessary for generating a detectable signal [37] [36].
- Sequencing by Synthesis (SBS): The flow cell is subjected to cycles of nucleotide incorporation. In the widely used Illumina platform, fluorescently labeled, reversible-terminator nucleotides are added one at a time. After each incorporation cycle, a high-resolution camera captures the color (corresponding to the base identity) of every cluster on the flow cell. The terminator group is then removed, allowing the next cycle to begin [37] [36] [38].
- Data Analysis and Alignment: The massive volume of short sequence reads generated (millions to billions) is processed by bioinformatics pipelines. These pipelines filter for quality and align the reads to a reference genome to identify mutations, such as indels, at the target site with single-nucleotide resolution [4] [37].
The core principle of NGS is its ability to simultaneously sequence millions of DNA fragments, providing unparalleled depth and sensitivity for detecting even rare editing events in a heterogeneous cell population [37] [38].
Diagram 2: NGS workflow and parallel sequencing principle.
Direct Comparative Analysis: Performance and Practicality
The choice between TIDE and NGS involves balancing multiple factors, including cost, depth of information, throughput, and required expertise. The following table provides a structured comparison based on these key parameters.
| Parameter | TIDE Assay | Next-Generation Sequencing (NGS) |
|---|---|---|
| Core Principle | Computational decomposition of Sanger sequencing traces [9] | Massively parallel sequencing of DNA fragments [37] [38] |
| Throughput | Low to medium; ideal for single or a few targets [4] | Very high; suitable for hundreds to thousands of targets or samples [4] [38] |
| Hands-on & Data Turnaround Time | ~2-3 days [25] | Several days to weeks (including library prep and data analysis) [4] [25] |
| Cost per Sample | Low (relies on standard PCR and Sanger sequencing) [25] | High (reagents, instrumentation, and bioinformatics) [4] [25] |
| Quantitative Accuracy | Good correlation with NGS for mid-range efficiencies (R² values high in validation studies) [4] | High; considered the "gold standard" for accuracy and sensitivity [4] [7] |
| Sensitivity (Variant Detection Limit) | ~5% variant allele frequency [4] | ~1% variant allele frequency or lower [38] |
| Information Detail | Identifies major indel types and their overall frequency; limited in complex mixtures [10] | Provides single-nucleotide resolution of all indels, including complex and large structural variants [4] [37] |
| Ease of Use / Bioinformatics Burden | Low; user-friendly web tool with instant analysis [4] [9] | High; requires specialized bioinformatics expertise and computational resources [4] [37] |
| Ideal Use Case | Rapid screening of sgRNA activity and preliminary efficiency checks [4] [8] | Comprehensive characterization of editing outcomes, detection of rare events, and therapeutic application validation [4] [38] |
Supporting Experimental Data and Validation
A landmark study directly compared CRISPR editing efficiency estimates from various methods with targeted NGS, which served as the benchmark. The study found that while the popular T7E1 mismatch assay often misrepresented editing efficiencies, both TIDE and NGS predicted similar editing efficiencies for pools of edited cells [7]. This validates TIDE as a quantitatively reliable method for determining overall indel frequency in bulk cell populations.
However, a more recent systematic comparison using artificial sequencing templates with predefined indels highlighted a limitation of TIDE and similar computational tools. The study reported that while these tools accurately estimate net indel sizes, their ability to deconvolute the exact sequences of complex indels can be variable and has certain limitations [10]. In such scenarios, the comprehensive sequence data provided by NGS becomes indispensable.
Essential Research Reagents and Solutions
Successful implementation of either TIDE or NGS for CRISPR analysis requires a foundational set of laboratory reagents and solutions. The following table details these key materials and their functions.
| Reagent / Solution | Function | Required in TIDE? | Required in NGS? |
|---|---|---|---|
| High-Fidelity DNA Polymerase | Accurate amplification of the target genomic locus from sample DNA to minimize PCR-introduced errors. | Yes [9] | Yes [37] |
| Sanger Sequencing Kit & Capillary Electrophoresis System | Generates the sequence chromatogram data file (.ab1) from the PCR amplicon for TIDE analysis. | Yes [9] [8] | No |
| TIDE Web Tool | The dedicated online software that performs the decomposition algorithm to quantify indels. | Yes [9] [25] | No |
| NGS Library Prep Kit | Contains enzymes and buffers to fragment DNA and ligate platform-specific adapter sequences. | No | Yes [37] [36] |
| NGS Platform-Specific Sequencing Kit | Contains the fluorescent nucleotides, buffers, and flow cells required for the sequencing-by-synthesis reaction. | No | Yes [37] [38] |
| Bioinformatics Software (e.g., BWA, GATK) | Tools for aligning sequence reads to a reference genome and calling genetic variants. | No | Yes [37] [38] |
The choice between TIDE and NGS is not a matter of which is universally superior, but which is optimal for a given research context.
TIDE is the recommended tool for rapid, low-cost screening during initial gRNA optimization and for projects where knowing the overall editing efficiency and broad categories of indels is sufficient. Its simplicity and speed make it ideal for labs without specialized bioinformatics support or for high-throughput preliminary screens [4] [25].
NGS is the unequivocal choice for comprehensive, high-resolution analysis. It is essential for clinical applications, therapeutic development, and basic research where precise sequence-level detail of every mutation is required. This includes detecting rare editing events, characterizing complex heterogeneous cell populations, and identifying unexpected outcomes like large deletions [4] [7].
A powerful and cost-effective strategy employed by many research groups is to use these methods in a complementary, two-stage process: using TIDE for initial, rapid screening of multiple gRNAs or experimental conditions, followed by confirmation and deep characterization of the most promising candidates with NGS. This hybrid approach leverages the strengths of both methods, balancing efficiency with comprehensiveness to accelerate genome editing research and development.
The rapid advancement of CRISPR-Cas9 genome editing technologies has created an urgent need for reliable methods to quantify editing efficiency. Among the various techniques available, Tracking of Indels by Decomposition (TIDE) has emerged as a popular, accessible tool that uses Sanger sequencing data to assess CRISPR-generated mutations. However, as the field matures, researchers must understand how this method compares to established gold-standard techniques, particularly when making critical decisions about guide RNA selection or validating editing efficiency for therapeutic applications. This guide provides an objective comparison of TIDE's performance against other quantification methods, drawing on recent benchmarking studies to help researchers select the most appropriate analytical approach for their specific needs.
Before examining TIDE's specific performance characteristics, it is essential to understand the landscape of available methods for quantifying CRISPR editing outcomes. These techniques generally fall into three categories: next-generation sequencing-based approaches, Sanger sequencing-based computational tools, and enzyme-based mismatch detection assays.
Table 1: Major CRISPR Editing Analysis Methods
| Method | Technology | Key Advantages | Primary Limitations |
|---|---|---|---|
| Targeted Amplicon Sequencing (AmpSeq) | Next-generation sequencing | High sensitivity and accuracy; comprehensive indel profiling [18] | Expensive; requires bioinformatics expertise; longer turnaround [4] |
| Inference of CRISPR Edits (ICE) | Sanger sequencing + computational analysis | Cost-effective; user-friendly interface; comparable to NGS [4] | Limited capability for complex indels or knock-in sequences [10] |
| Tracking of Indels by Decomposition (TIDE) | Sanger sequencing + computational analysis | Simple workflow; rapid results; cost-effective [1] [25] | Cannot detect large deletions; accuracy declines with complex edits [1] [10] |
| T7 Endonuclease I (T7E1) | Mismatch cleavage assay | Lowest cost; technically simple; fast results [4] | Semi-quantitative; low dynamic range; no sequence information [4] [7] |
| Droplet Digital PCR (ddPCR) | Probe-based quantification | Absolute quantification; high precision; sensitive [18] [8] | Requires specific probe design; limited to predefined edits [8] |
| Indel Detection by Amplicon Analysis (IDAA) | Capillary electrophoresis | Size-based indel detection; moderate throughput [7] | No sequence-level information; limited resolution [7] |
The TIDE method specifically analyzes Sanger sequencing chromatograms from both control and edited samples, using a decomposition algorithm to quantify the spectrum and frequency of insertions and deletions (indels). The web-based tool requires two standard capillary sequencing reactions and provides quantitative data on editing efficiency within a simple, accessible interface [1] [25].
Experimental Protocols for Key Methods
TIDE Assay Protocol
The TIDE methodology follows a straightforward workflow requiring standard molecular biology reagents [1] [25]:
PCR Amplification: Amplify the target region from both control (non-edited) and experimental (edited) genomic DNA samples using a single pair of primers. The target region should be approximately 700 bp, with the CRISPR cut site preferably located about 200 bp downstream from the sequencing start site.
Sanger Sequencing: Perform standard capillary sequencing of the PCR products. The sequencing should cover a stretch of DNA enclosing the designed editing site. ABIF (.ab1) or SCF (.scf) file formats are supported.
Online Analysis:
- Upload the sequencing files to the TIDE web tool (http://shinyapps.datacurators.nl/tide/)
- Input the 20nt sgRNA sequence (without PAM) immediately upstream of the PAM sequence
- Set parameters including alignment window (default: 100 bp upstream of break site), decomposition window (automatically set to maximum possible), indel size range (default: 10 bp), and p-value threshold (default: 0.001)
- The algorithm decomposes the mixed sequencing traces and quantifies indel frequencies
Figure 1: TIDE assay workflow showing the six key steps from PCR amplification to results output.
Targeted Amplicon Sequencing Protocol
The gold standard AmpSeq protocol provides a benchmark for comparison [18]:
PCR Amplification: Design primers flanking the target site and amplify with barcoded primers to enable multiplexing.
Library Preparation: Purify PCR products and quantify using fluorometric methods. Prepare sequencing libraries using commercial kits compatible with Illumina platforms.
Sequencing: Perform paired-end sequencing (e.g., 2×250 bp) on Illumina MiSeq or similar platforms.
Bioinformatic Analysis:
- Demultiplex sequences based on barcodes
- Align reads to reference sequence using tools like CRISPResso2 or custom pipelines
- Quantify indel frequencies and spectra
T7E1 Assay Protocol
As a commonly used but less quantitative method, T7E1 provides a useful comparison point [4] [7]:
PCR Amplification: Amplify the target region from both control and edited samples.
Heteroduplex Formation:
- Denature and reanneal PCR products by heating to 95°C followed by slow cooling to room temperature
- This forms heteroduplexes between wild-type and mutated strands
T7E1 Digestion:
- Incubate reannealed DNA with T7 Endonuclease I at 37°C for 30 minutes
- The enzyme cleaves mismatched DNA at heteroduplex sites
Gel Electrophoresis:
- Separate digestion products by agarose gel electrophoresis
- Quantify band intensities to estimate editing efficiency
Comparative Performance Analysis
Quantitative Accuracy Assessment
Recent benchmarking studies have systematically evaluated the accuracy of TIDE compared to other methods, with targeted amplicon sequencing (AmpSeq) serving as the reference standard.
Table 2: Quantitative Comparison of Editing Efficiency Measurements Across Methods
| Method | Correlation with AmpSeq | Dynamic Range | Sensitivity for Low-Frequency Edits | Complex Indel Detection |
|---|---|---|---|---|
| TIDE | R² = 0.65-0.90 [18] [10] | 10-90% editing [10] | Limited below 5-10% [10] | Limited for complex patterns [10] |
| ICE | R² = 0.96 vs NGS [4] | 5-95% editing [4] [10] | Moderate (~5%) [10] | Better than TIDE [4] |
| T7E1 | Poor correlation [7] | 10-30% effective range [7] | Insensitive below 10% [7] | Cannot detect specific sequences [4] |
| ddPCR | R² > 0.95 [18] [8] | 0.1-100% [8] | High (0.1-1%) [8] | Limited to predefined edits [8] |
| PCR-CE/IDAA | R² > 0.90 [18] | 5-95% [18] | Moderate (~5%) [18] | Size-based only [18] |
A comprehensive 2025 benchmarking study evaluated these methods across 20 sgRNA targets in plants and found that while TIDE provided reasonable estimates for mid-range editing efficiencies (20-70%), it showed significant variability at the extremes of low (<10%) and high (>80%) editing efficiency [18]. The study also noted that base-calling algorithms used in Sanger sequencing can significantly impact TIDE's sensitivity, with PeakTrace base-caller potentially underestimating low-frequency edits [18].
Detection of Diverse Editing Outcomes
Different CRISPR analysis methods vary significantly in their ability to detect various types of editing outcomes beyond simple indels.
Table 3: Capabilities for Detecting Different Editing Outcomes
| Editing Outcome | TIDE | ICE | AmpSeq | T7E1 | ddPCR |
|---|---|---|---|---|---|
| Small indels (<10 bp) | Excellent [1] | Excellent [4] | Excellent [18] | Good [4] | Targeted only [8] |
| Large deletions (>100 bp) | Cannot detect [1] | Limited [4] | Excellent [18] | Cannot detect [39] | Targeted only [40] |
| Complex rearrangements | Cannot detect [1] | Limited [4] | Excellent [39] | Cannot detect [39] | Targeted only [40] |
| Precise knock-ins | Limited [10] | Limited [10] | Excellent [18] | Cannot detect [4] | Excellent [8] |
| Unresolved DSBs | Cannot detect [40] | Cannot detect [40] | Limited [40] | Cannot detect [40] | Good [40] |
A critical limitation of TIDE is its inability to detect large deletions, which are now recognized as a common outcome of CRISPR editing. As noted in a 2025 study, "The method will not capture megabase-long deletions that can originate by CRISPR/Cas9 induced DSB" [1]. This represents a significant blind spot, as large deletions and complex rearrangements may have substantial functional consequences in edited cells.
Method-Specific Limitations and Biases
Each quantification method carries specific limitations that can impact data interpretation:
TIDE-Specific Limitations:
- Requires high-quality Sanger sequencing with the break site approximately 200bp downstream from sequencing start [1]
- Accuracy decreases with poor sequencing quality or when the editing site is too close to sequencing primers [1]
- Cannot resolve complex mixtures of indels effectively [10]
- Assumes DSB occurs between nucleotides 17 and 18 of the sgRNA sequence [1]
Comparative Method Limitations:
- T7E1: Dramatically underestimates high-efficiency editing (>30%) and overestimates low-efficiency editing [7]
- AmpSeq: PCR amplification bias can affect quantification accuracy [40]
- IDAA: Provides no sequence information, only fragment sizes [7]
Research Reagent Solutions
Table 4: Essential Research Reagents for CRISPR Editing Analysis
| Reagent/Resource | Function | Example Suppliers/Platforms |
|---|---|---|
| TIDE Web Tool | Computational analysis of Sanger sequencing data | https://tide.nki.nl [1] |
| ICE Web Tool | Alternative computational analysis platform | https://ice.synthego.com [4] |
| T7 Endonuclease I | Mismatch detection enzyme for T7E1 assay | New England Biolabs [8] |
| Q5 Hot Start High-Fidelity Master Mix | PCR amplification for sequencing | New England Biolabs [8] |
| Sanger Sequencing Services | Capillary sequencing for TIDE input | Macrogen, Eurofins [8] |
| ddPCR Systems | Absolute quantification of editing events | Bio-Rad [18] |
Applications and Recommendations
When to Use TIDE
TIDE represents an optimal choice for researchers needing rapid, cost-effective analysis of editing efficiency when:
- Screening multiple sgRNAs during initial optimization phases
- Budget constraints preclude NGS approaches
- Simple indel spectra are expected without large deletions
- Editing efficiency falls in the mid-range (10-80%)
- Rapid turnaround is prioritized over comprehensive characterization [4] [25]
When to Choose Alternative Methods
Gold-Standard AmpSeq is recommended when:
- Comprehensive characterization of editing outcomes is required
- Detecting large deletions or complex rearrangements is essential
- Highest sensitivity and accuracy are needed for publication or therapeutic development
- Resources and bioinformatics expertise are available [18] [39]
ddPCR is preferable for:
- Absolute quantification of specific known edits
- Detecting low-frequency editing events (<5%)
- Applications requiring high precision and reproducibility [18] [8]
T7E1 may suffice for:
- Preliminary screening when cost is the primary concern
- Qualitative assessment of editing presence/absence
- Situations where sequence-level resolution is unnecessary [4]
Figure 2: Decision tree for selecting appropriate CRISPR analysis methods based on research needs and constraints.
TIDE provides a valuable balance of accessibility, cost-effectiveness, and reasonable accuracy for many CRISPR editing applications. Its performance is substantially superior to simple enzymatic methods like T7E1 and approaches the accuracy of NGS-based methods for standard editing scenarios involving small indels. However, researchers must recognize its limitations in detecting large deletions and complex rearrangements, which may represent important safety considerations in therapeutic applications. For comprehensive characterization of editing outcomes, particularly in clinical or precise genome editing applications, targeted amplicon sequencing remains the gold standard, with TIDE serving as an effective tool for initial screening and optimization phases. As CRISPR technologies continue to evolve, method selection should be guided by the specific research questions, regulatory requirements, and resources available.
The emergence of CRISPR-based genome editing has revolutionized biological research, enabling precise genetic modifications across a wide range of organisms and cell types [18]. However, the efficiency and outcomes of editing experiments vary significantly depending on multiple factors, including guide RNA (gRNA) design, delivery method, and cellular context [16] [41]. Accurate quantification of editing efficiency is therefore paramount for developing robust genome editing applications, from functional genomics to therapeutic development [18] [8].
Various methods have been developed to detect and quantify insertion and deletion (indel) mutations resulting from non-homologous end joining (NHEJ) repair. These techniques range from simple enzyme-based assays to sophisticated sequencing approaches, each with distinct strengths, limitations, and resource requirements [8] [4]. This guide provides a comprehensive, data-driven comparison of the most widely used methods, focusing on their performance characteristics, technical requirements, and suitability for different experimental scenarios, with particular emphasis on the Tracking of Indels by Decomposition (TIDE) assay within the broader context of genome editing analysis.
Comparative Performance Analysis of Genome Editing Assessment Methods
Quantitative Method Comparison
Extensive benchmarking studies have systematically evaluated the performance of different genome editing analysis techniques. The table below summarizes key characteristics based on experimental comparisons.
Table 1: Performance characteristics of major genome editing assessment methods
| Method | Reported Sensitivity Range | Key Strengths | Significant Limitations | Cost & Accessibility |
|---|---|---|---|---|
| TIDE | Good for simple indels [10] | Rapid (1-2 days); cost-effective; simple workflow requiring only PCR and Sanger sequencing [3] [13] | Limited detection of large indels (>50 bp); accuracy decreases with complex indel patterns [10] [5] | Low; web tool freely available [3] |
| ICE | Comparable to NGS (R²=0.96) [4] | User-friendly interface; detects large indels; provides knockout score focusing on frameshifts [4] | Performance varies with editing complexity [10] | Low; web tool freely available |
| T7E1 | Low; fails to detect edits <10% and underestimates high efficiency [41] | Cheapest and fastest option; minimal equipment needs [4] | Semi-quantitative; no sequence information; highly variable results [8] [41] | Very low; requires only basic lab equipment |
| ddPCR | High; precise quantification [8] | Excellent accuracy and sensitivity; absolute quantification without standards [18] [8] | Requires specialized equipment and optimized probe design | High; requires specialized instrument |
| AmpSeq | Highest; considered "gold standard" [18] [41] | Most comprehensive and sensitive method; provides complete sequence-level data [18] [4] | Expensive; long turnaround; requires bioinformatics expertise [18] [4] | Highest; requires sequencing platform and bioinformatics resources |
Experimental Validation Data
Independent studies have quantified the performance relationships between these methods. When compared to amplicon sequencing (AmpSeq) as a benchmark, PCR-capillary electrophoresis/IDAA and ddPCR methods demonstrate high accuracy [18]. However, significant discrepancies have been observed between methods, particularly with the T7E1 assay, which often shows poor correlation with sequencing-based approaches [41]. One study found that sgRNAs with apparently similar activity by T7E1 (both ~28%) actually had dramatically different true editing efficiencies when measured by next-generation sequencing (40% vs. 92%) [41].
For Sanger sequencing-based computational tools, a systematic comparison using artificial sequencing templates with predetermined indels revealed that all tools (TIDE, ICE, and DECODR) performed with acceptable accuracy for simple indels with few base changes [10]. However, as editing patterns became more complex, the estimated values showed greater variability between tools [10]. Among these tools, DECODR provided the most accurate estimations of indel frequencies for the majority of samples, though each tool has specific optimal use cases [10].
Decision Matrix for Method Selection
Experimental Workflow and Resource Considerations
The selection of an appropriate genome editing assessment method depends on multiple experimental factors. The following decision matrix integrates performance data with practical research constraints to guide method selection.
Diagram 1: Method selection workflow (47 characters)
Application-Specific Recommendations
Rapid Screening of Multiple gRNAs: For initial testing of numerous gRNAs during optimization phases, TIDE and ICE provide the optimal balance of information content, cost, and throughput [4]. These methods significantly outperform T7E1 in accuracy while avoiding the resource-intensive nature of AmpSeq [41].
Validation of Therapeutic Edits: For clinical applications or precise editing quantification, AmpSeq remains the gold standard, with ddPCR serving as a highly accurate alternative for well-characterized edits [18] [8]. The comprehensive sequence information provided by AmpSeq is essential for characterizing complex editing outcomes in heterogeneous cell populations [18].
Low-Resource Settings: When funding or expertise is limited, and only basic editing confirmation is needed, the T7E1 assay provides a functional though limited option [4]. However, researchers should recognize its significant limitations in quantitative accuracy and sensitivity [41].
Systems Prone to Large Deletions: In experimental systems where large deletions (>50 bp) are common, ICE or the newer PtWAVE tool (which detects indels up to 200 bp) are recommended over TIDE, which has limited detection range for larger indels [5].
Detailed Experimental Protocols
TIDE Assay Workflow
The TIDE method provides a straightforward protocol for quantifying indel frequencies [13]:
Table 2: Key research reagents for TIDE assay
| Reagent/Equipment | Specification | Function in Protocol |
|---|---|---|
| Genomic DNA Isolation Kit | Standard commercial kit | Extract high-quality DNA from at least 1000 edited cells |
| PCR Primers | Flanking target site (200 bp from cut site) | Amplify 500-1500 bp region surrounding editing site |
| PCR Master Mix | Standard Taq polymerase with buffer | Amplify target region from genomic DNA |
| Agarose Gel Electrophoresis | 1-2% agarose gel | Confirm PCR product quality and specificity |
| PCR Purification Kit | Standard commercial kit | Purify PCR products before sequencing |
| Sanger Sequencing | Commercial service or core facility | Generate sequence traces in .ab1 format |
| TIDE Web Tool | https://tide.nki.nl | Analyze sequencing traces and quantify indels |
Procedure:
DNA Extraction: Isolate genomic DNA from edited cells 3-4 days post-transfection using a standard genomic DNA isolation kit. A minimum of 1000 cells should be processed to adequately sample mutation complexity [13].
PCR Amplification: Design primers to amplify a 500-1500 bp region surrounding the target site, with the Cas9 cut site positioned approximately 200 bp downstream from the sequencing start site [13]. Perform PCR with standard conditions: initial denaturation at 95°C for 1 minute, followed by 25-30 cycles of denaturation (95°C for 15 seconds), annealing (55-58°C for 15 seconds), and extension (72°C for 10 seconds per kb) [13].
Product Verification: Analyze 5-10% of the PCR product on a 1-2% agarose gel to confirm a single, sharp band of expected size [13].
PCR Purification: Purify the remaining PCR product using a commercial PCR purification kit according to manufacturer's instructions [13].
Sanger Sequencing: Submit purified PCR products for Sanger sequencing using the same primer as in step 2. Ensure sequence trace files are saved in .ab1 or .scf format for TIDE analysis [13].
TIDE Analysis: Upload the sequencing trace files from both control (unedited) and experimental (edited) samples to the TIDE web tool (https://tide.nki.nl). The algorithm will decompose the mixed sequences and provide quantitative indel frequency data [3] [13].
T7E1 Assay Protocol
For comparative purposes, the T7E1 assay protocol is summarized below:
PCR Amplification: Amplify the target region as described in the TIDE protocol [8].
Heteroduplex Formation: Denature and reanneal PCR products by heating to 95°C for 5-10 minutes, then slowly cooling to 25°C (0.1°C/second) [8] [41].
T7E1 Digestion: Digest the heteroduplexed DNA with T7 Endonuclease I (2 units) in appropriate buffer at 37°C for 30 minutes [8].
Analysis: Separate digestion products by agarose gel electrophoresis (1-2% gel). Cleavage products indicate the presence of edited alleles [8] [41].
Quantification: Estimate editing efficiency by measuring band intensities using densitometry software with the formula: % indel = 100 × (1 - [1 - (a + b)/(a + b + c)]^{1/2}), where c is the integrated intensity of the undigested PCR product, and a and b are the integrated intensities of the cleavage products [41].
Advanced Applications and Specialized Scenarios
TIDER for Templated Editing
For experiments involving homology-directed repair (HDR) with donor templates, TIDER (Tracking of Insertions, DEletions, and Recombination events) extends TIDE functionality to quantify precise template-directed mutations against a background of random indels [3] [13]. The TIDER method requires an additional reference DNA sample, which can be prepared using a two-step PCR protocol that incorporates the desired mutation into an amplicon, serving as a reference for the decomposition algorithm [3] [13].
Detection of Large Deletions
Conventional TIDE analysis struggles to detect large deletions exceeding 50 bp [5]. Recent algorithmic improvements have addressed this limitation. The PtWAVE tool, for example, extends detection capabilities to indels up to 200 bp through progressive adjustment of estimated mutation sequence patterns and consideration of Bayesian information criterion [5]. This is particularly valuable for systems where large deletions frequently occur following nuclease activity.
The selection of an appropriate genome editing assessment method requires careful consideration of experimental needs, resource constraints, and required data quality. While AmpSeq remains the gold standard for comprehensive analysis, TIDE and ICE provide excellent alternatives for most research applications, balancing cost, throughput, and information content. The T7E1 assay, despite its limitations, may suffice for basic editing confirmation when resources are severely constrained. As genome editing applications continue to expand into more complex biological systems and therapeutic development, appropriate validation methods will remain crucial for generating reliable, reproducible results. By applying the decision matrix and experimental protocols outlined in this guide, researchers can make informed choices about the most suitable quantification method for their specific genome editing applications.
Conclusion
The TIDE assay remains a powerful, cost-effective, and accessible method for the initial quantification and characterization of indel profiles in genome-edited cell pools. Its strength lies in providing rapid, quantitative data on editing efficiency and indel spectra from standard Sanger sequencing, making it an invaluable tool for researchers optimizing CRISPR systems. However, its limitations in detecting large deletions and templated mutations necessitate complementary methods like TIDER or NGS for comprehensive analysis. As genome editing advances toward clinical applications, the principles of efficient, reproducible validation that TIDE embodies will only grow in importance. Future developments in decomposition algorithms, such as those seen in PtWAVE for larger indel detection, promise to enhance these accessible tools further, solidifying their role in the scalable validation required for therapeutic genome editing.
References
- 1. TIDE: Tracking of Indels by DEcomposition [https://apps.datacurators.nl/tide/]
- 2. Systematic Comparison of Computational Tools for Sanger ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10854981/]
- 3. TIDE [https://tide.nki.nl/]
- 4. How to Pick the Best CRISPR Data Analysis Method for ... [https://www.synthego.com/guide/how-to-use-crispr/analysis-methods/]
- 5. PtWAVE: a high-sensitive deconvolution software of ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06139-8]
- 6. Discrepancies in indel software resolution with somatic ... [https://www.nature.com/articles/s41598-023-41109-1]
- 7. A Survey of Validation Strategies for CRISPR-Cas9 Editing [https://www.nature.com/articles/s41598-018-19441-8]
- 8. Comparative Analysis of Methods for Assessing On-Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11932317/]
- 9. Easy quantitative assessment of genome editing by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4267669/]
- 10. Systematic Comparison of Computational Tools for Sanger ... [https://www.mdpi.com/2073-4409/13/3/261]
- 11. Tracy: basecalling, alignment, assembly and deconvolution of ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6635-8]
- 12. SuperDecode: An integrated toolkit for analyzing mutations ... [https://www.sciencedirect.com/science/article/pii/S1674205225000929]
- 13. Rapid Quantitative Evaluation of CRISPR Genome Editing ... [https://link.springer.com/protocol/10.1007/978-1-4939-9170-9_3]
- 14. A Comparison of Techniques to Evaluate the Effectiveness ... [https://www.sciencedirect.com/science/article/abs/pii/S016777991730272X]
- 15. A Bioinformatic Workflow for InDel Analysis in the Wheat ... [https://www.mdpi.com/1422-0067/22/23/13076]
- 16. Optimization of gene knockout approaches and sgRNA ... [https://www.nature.com/articles/s41598-025-24184-4]
- 17. Validating CRISPR: How to Confirm Successful Editing [https://bitesizebio.com/47275/validate-crispr-experiment/]
- 18. Comprehensive benchmarking of genome editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12144422/]
- 19. PCR for Sanger Sequencing [https://www.thermofisher.com/us/en/home/life-science/sequencing/sanger-sequencing/sanger-dna-sequencing/pcr-sanger-sequencing.html]
- 20. Sanger Sequencing Best Practices - A guide to sample ... [https://genome.center.uconn.edu/cgi-services/sequencing-best-practices-a-guide-to-sample-preparation-and-troubleshooting/]
- 21. Sample Preparation - GENEWIZ from Azenta Life ... [https://www.genewiz.com/public/resources/sample-submission-guidelines/sanger-sequencing-sample-submission-guidelines/sample-preparation]
- 22. A Beginner's Guide to Sanger Sequencing Results... [https://geneticeducation.co.in/a-beginners-guide-to-sanger-sequencing-results/]
- 23. Comparative Analysis of Methods for Assessing On-Target ... [https://www.mdpi.com/2409-9279/8/2/23]
- 24. Gene editing frequencies determined by TIDER and TIDE [https://bio-protocol.org/exchange/minidetail?id=7902335&type=30]
- 25. TIDE [https://www.nki.nl/research/research-groups/bas-van-steensel/our-technologies/tide]
- 26. Rapid Quantitative Evaluation of CRISPR Genome Editing ... [https://pubmed.ncbi.nlm.nih.gov/30912038/]
- 27. CRISPR 101: Validating Your Genome Edit [https://blog.addgene.org/crispr-101-validating-your-genome-edit]
- 28. analyzing TIDE results [https://groups.google.com/g/crispr/c/mXiZaIiRZ4Y]
- 29. Mastering DNA chromatogram analysis in Sanger ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10643650/]
- 30. Easy quantification of template-directed CRISPR/Cas9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6007333/]
- 31. R² in Survey Analysis: What It Is and Why It Matters [https://blog.polling.com/r%C2%B2-in-survey-analysis-what-it-is-and-why-it-matters/]
- 32. Detection and quantification of unintended large on-target ... [https://www.sciencedirect.com/science/article/abs/pii/S246845112300034X]
- 33. PtWAVE: a high-sensitive deconvolution software of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12039204/]
- 34. How To Use ICE: A Detailed Guide for Analyzing CRISPR ... [https://www.synthego.com/guide/how-to-use-crispr/ice-analysis-guide/]
- 35. Case study: streamlining routine CRISPR validation with ... [https://nanoporetech.com/resource-centre/case-study-streamlining-routine-crispr-validation-with-oxford-nanopore-sequencing]
- 36. Next-generation sequencing: 2025 Revolution [https://lifebit.ai/blog/next-generation-sequencing/]
- 37. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 38. Next-Generation Sequencing: A Review of Its Transformative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12523276/]
- 39. Verification of CRISPR editing and finding transgenic ... [https://www.sciencedirect.com/science/article/pii/S104620232100027X]
- 40. Unveiling the cut-and-repair cycle of designer nucleases in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12583642/]
- 41. A Survey of Validation Strategies for CRISPR-Cas9 Editing | Scientific... [https://www.nature.com/articles/s41598-018-19441-8?error=cookies_not_supported&code=eb36e8b9-f67d-4144-b15a-a6864a812e84]