QIIME vs. Mothur: A Comprehensive Guide to Choosing and Optimizing Your 16S rRNA Bioinformatics Pipeline
This article provides a foundational to advanced overview of the two predominant bioinformatics pipelines, QIIME and mothur, for 16S rRNA amplicon data analysis.
QIIME vs. Mothur: A Comprehensive Guide to Choosing and Optimizing Your 16S rRNA Bioinformatics Pipeline
Abstract
This article provides a foundational to advanced overview of the two predominant bioinformatics pipelines, QIIME and mothur, for 16S rRNA amplicon data analysis. Tailored for researchers, scientists, and drug development professionals, it explores the core principles and appropriate use-cases for each tool. The content delivers detailed methodological workflows, addresses common troubleshooting and optimization challenges, and synthesizes evidence from recent comparative studies to validate pipeline selection. By integrating practical guidance with current research findings, this guide aims to empower users to generate robust, reproducible, and biologically meaningful microbiome data for biomedical and clinical applications.
Understanding 16S rRNA Analysis and the Key Players: QIIME and Mothur
16S ribosomal RNA (rRNA) gene sequencing is a cornerstone molecular method for identifying and classifying bacteria and archaea within complex biological samples. The 16S rRNA gene is approximately 1500 base pairs long and contains nine hypervariable regions (V1-V9) interspersed between conserved regions. This genetic structure makes it an ideal target for phylogenetic studies: the conserved regions enable amplification with universal primers, while the variable regions provide the sequence diversity necessary for taxonomic differentiation [1] [2].
This culture-free approach has revolutionized microbial ecology by allowing researchers to characterize microbial communities that are difficult or impossible to study using traditional laboratory cultivation methods. In biomedical research, 16S rRNA sequencing provides insights into the composition of human-associated microbiota and their roles in health and disease, making it fundamental to understanding the human microbiome [3] [1].
Sequencing Technology Comparisons
The selection of sequencing technology significantly influences the scope and resolution of a 16S rRNA study. The table below compares the key characteristics of the primary platforms used in modern research.
Table 1: Comparison of 16S rRNA Gene Sequencing Platforms
| Sequencing Platform | Read Type | Targeted Region | Typical Taxonomic Resolution | Key Advantages | Primary Considerations |
|---|---|---|---|---|---|
| Illumina MiSeq | Short-read (∼300 bp) | Single hypervariable regions (e.g., V3-V4) | Genus-level | High accuracy (Q30+), low per-read cost | Limited resolution for closely related species |
| Oxford Nanopore (ONT) | Long-read (Full-length) | V1-V9 (∼1500 bp) | Species-level | Real-time sequencing, portable devices, lower instrument cost | Higher raw error rate, though improved with recent chemistry [4] |
| PacBio SMRT | Long-read (Full-length) | V1-V9 (∼1500 bp) | Species-level | High-fidelity (HiFi) reads, high single-read accuracy | Higher cost per sample for equivalent read depth [5] |
Multiple studies have demonstrated that full-length 16S rRNA sequencing improves species-level classification. One study on human microbiome samples showed that while Illumina (V3-V4) and PacBio (V1-V9) assigned a similar percentage of reads to the genus level (∼95%), PacBio assigned a significantly higher proportion to the species level (74.14% vs. 55.23%) [5]. Similarly, Oxford Nanopore's full-length sequencing has been shown to identify more specific bacterial biomarkers for conditions like colorectal cancer compared to Illumina's V3-V4 approach [4].
Experimental Protocol: From Sample to Data
A standard 16S rRNA gene sequencing workflow involves multiple critical steps, from sample collection to sequencing, each of which can impact the final results.
Sample Preparation and DNA Extraction
- Sample Collection: Samples (e.g., feces, saliva, tissue) must be collected with strict adherence to sterility to prevent contamination. Immediate freezing at -20°C or -80°C is crucial to preserve microbial composition. For temporary storage, samples can be held at 4°C or in preservation buffers [1].
- DNA Extraction: This process involves cell lysis (chemical and mechanical), precipitation of DNA using salt and alcohol, and final purification to remove contaminants. The choice of DNA extraction kit should be appropriate for the sample type, as this can significantly impact sequencing results [1].
Library Preparation and Sequencing
- PCR Amplification: The extracted DNA is amplified using primers that target specific hypervariable regions of the 16S rRNA gene. The choice of primer pair and the targeted region (e.g., V3-V4 vs. V1-V9) is a key experimental decision that influences which taxa are detected [1] [5].
- Barcoding and Clean-up: For multiplexing, unique molecular barcodes are added to samples during a second PCR. The amplified DNA is then cleaned, typically with magnetic beads, to remove impurities and select for the correct fragment size [1].
- Sequencing: The final library is loaded onto a sequencer (Illumina, Nanopore, or PacBio) for high-throughput analysis [1].
The following diagram illustrates the complete wet-lab workflow:
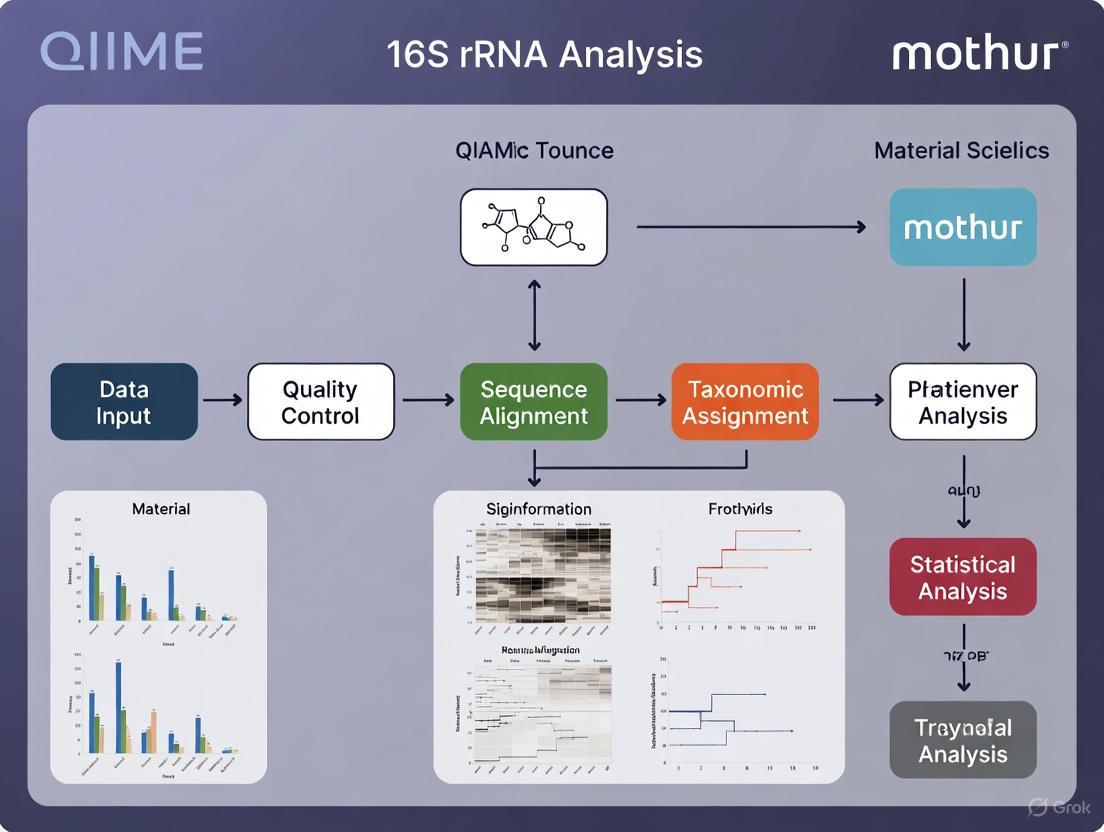
Bioinformatics Pipelines for Data Analysis
The raw sequencing data must be processed through a bioinformatics pipeline to generate actionable biological insights. Two of the most established pipelines are QIIME and mothur.
Both QIIME and mothur follow a similar core workflow for processing 16S rRNA amplicon data, though their underlying implementations and philosophies differ.
QIIME vs. Mothur: A Comparative Analysis
The choice between QIIME and mothur can influence results, particularly for low-abundance taxa.
Table 2: Comparison of QIIME and Mothur Bioinformatics Pipelines
| Feature | QIIME | Mothur |
|---|---|---|
| Primary Language | Python (wrapper for external tools) [6] | C/C++ (standalone compiled code) [6] |
| Development Strategy | Integrates multiple independent tools [6] | Self-contained; most tools reimplemented in C++ [6] |
| Installation | Can be complex due to dependencies [6] | Straightforward; single executable [6] |
| Performance | Slower for computationally intensive tasks (e.g., alignment) [6] | Faster execution for core algorithms (e.g., 21.9x faster alignment) [6] |
| Database Influence | With GreenGenes, assigned fewer OTUs and genera for RA < 10% [7] [8] | With GreenGenes, higher richness, more genera detected for RA < 10% [7] [8] |
| Recommendation | SILVA database attenuates differences between pipelines [7] [8] | SILVA database is preferred for comparable results with QIIME [7] [8] |
A study on rumen microbiota found that while both tools showed a high correlation (>0.99) for the relative abundance of major genera, mothur detected a larger number of Operational Taxonomic Units (OTUs) and genera, especially for low-abundance microorganisms (relative abundance < 10%) when using the GreenGenes database. These differences can significantly impact beta diversity metrics. However, using the SILVA reference database attenuated these discrepancies, making the outputs of QIIME and mothur more comparable [7] [8].
Essential Research Reagents and Materials
Successful execution of a 16S rRNA sequencing experiment requires careful selection of reagents and reference databases.
Table 3: Key Research Reagent Solutions for 16S rRNA Gene Sequencing
| Item | Function/Description | Examples & Considerations |
|---|---|---|
| DNA Extraction Kit | Isolates microbial genomic DNA from complex samples. | Choice critical for low-biomass samples (e.g., skin swabs). Kits from Molzym GmbH & Co. KG used in clinical samples [9]. |
| PCR Primers | Amplifies the target hypervariable region of the 16S rRNA gene. | Region selection (e.g., V3V4, V1V9) influences results. Examples: 27F and 1492R for full-length [5]. |
| Sequencing Platform | Determines read length, accuracy, and cost. | Illumina for short-read; Oxford Nanopore or PacBio for full-length [4] [5]. |
| Reference Database | Essential for taxonomic classification of sequence reads. | SILVA and GreenGenes are common; database choice greatly influences identified species [7] [4]. |
| Bioinformatics Tools | Process raw sequences into taxonomic and ecological data. | QIIME and mothur are standard pipelines; DADA2 (Illumina) and Emu (ONT) for denoising [4] [10]. |
Applications in Biomedical Research
The application of 16S rRNA sequencing in biomedicine is vast and growing, providing insights into disease diagnostics, forensic science, and therapeutic development.
- Infectious Disease Diagnostics: 16S sequencing is a powerful tool for identifying pathogens in culture-negative clinical samples, especially after antibiotic administration. Next-Generation Sequencing (NGS) outperforms Sanger sequencing in polymicrobial samples, with one study showing a higher positivity rate (72% vs. 59%) and better detection of mixed infections [9].
- Forensic Identification: The human microbiome is unique enough to serve as a "microbial fingerprint." 16S rRNA gene sequencing, combined with machine learning, can match skin, oral, or soil microbiomes to individuals or specific locations with high accuracy, providing evidence for criminal investigations [3].
- Disease Biomarker Discovery: By comparing microbiomes between healthy and diseased individuals, researchers can identify specific microbial biomarkers. For example, full-length 16S sequencing with Oxford Nanopore has identified species like Fusobacterium nucleatum and Bacteroides fragilis as precise biomarkers for colorectal cancer, improving predictive models for the disease [4].
- Therapeutic Monitoring: This technology can track changes in a patient's microbiome in response to interventions such as antibiotics, probiotics, or dietary changes, enabling personalized treatment strategies.
In the field of microbial ecology, the analysis of 16S rRNA gene amplicon sequencing data has become a fundamental methodology for profiling complex microbial communities. Among the bioinformatic tools available, QIIME (Quantitative Insights Into Microbial Ecology) and mothur have emerged as two of the most prominent and widely adopted ecosystems [7] [11]. Since their initial releases within months of each other in 2009-2010, these pipelines have supported thousands of microbiome studies across diverse environments, from the human gut to industrial bioreactors [6]. Despite addressing similar analytical challenges, they embody fundamentally different philosophical approaches to software design, implementation, and user interaction. This article explores the core philosophies of these two ecosystems, provides structured comparisons of their performance, and offers practical guidance for researchers navigating the choice between them within modern bioinformatics workflows for 16S rRNA data analysis.
Philosophical Foundations and Architectural Design
Core Design Philosophies
The divergence between QIIME and mothur begins at the architectural level, reflecting distinct priorities in software design:
mothur adopts a unified toolset approach, implemented primarily in C++ to create a standalone, optimized executable. This design prioritizes computational efficiency, independence from external dependencies, and a consistent user experience through an integrated command-line interface [6]. As noted by its developers, "When you run a function from within mothur, you are running mothur code" [6]. This self-contained nature ensures stability and reduces installation complications, though it may limit community code contributions due to the C++ implementation barrier.
QIIME (particularly the contemporary QIIME 2 platform) embraces a modular, framework-based philosophy. Built primarily in Python, it functions as a "big wrapper that helps users to transition data between independent packages" [6]. This plugin-based architecture encourages community development and method integration, allowing specialized tools to be incorporated with "light wrapper" code while maintaining their original implementations [6]. The platform emphasizes data provenance tracking, reproducibility, and extensibility, with a focus on creating a transparent analytical record that documents every processing step [12] [13].
Implementation and Performance Characteristics
The choice of programming language profoundly influences performance characteristics and user interaction patterns:
Table 1: Implementation Characteristics of mothur and QIIME
| Aspect | mothur | QIIME/QIIME 2 |
|---|---|---|
| Primary Language | C/C++ (compiled) | Python (interpreted) |
| Execution Speed | Faster execution for core algorithms | Slower for computationally intensive tasks |
| Dependencies | Self-contained, minimal dependencies | Multiple external dependencies |
| Installation | Straightforward (single executable) | More complex (dependency management) |
| Extensibility | Limited by core development team | High (plugin architecture) |
| Provenance Tracking | Limited | Comprehensive (core feature) |
As observed in benchmarking studies, "Because of our overall development strategy we have worked very hard to make mothur a standalone software package. When you download mothur, you have mothur. All of it. You don't have to chase down external dependencies or worry about software licenses" [6]. This integrated approach translates to performance advantages for certain computationally intensive tasks, with mothur's aligner performing 21.9-times faster than QIIME's Python-based alternative in one comparison [6].
Conversely, QIIME 2's framework approach has facilitated ongoing innovation, with regular releases adding functionality such as cryptographic signing of results [14], enhanced visualization capabilities [12], and improved reporting features [14]. The provenance system automatically records all analytical steps, parameters, and computational environments, addressing critical reproducibility challenges in bioinformatics [13].
Comparative Performance Benchmarking
Taxonomic Composition and Diversity Assessment
Multiple studies have directly compared the analytical outcomes of QIIME and mothur pipelines using both mock communities and real biological samples. The choice between these platforms can influence the resulting biological interpretations, particularly for low-abundance taxa.
A comprehensive comparison using rumen microbiota samples found that both tools showed a high degree of agreement in identifying the most abundant genera (Bifidobacterium, Butyrivibrio, Methanobrevibacter, Prevotella, and Succiniclasticum), regardless of the reference database used [7] [15]. However, significant differences emerged for less abundant microorganisms (relative abundance < 10%), with mothur assigning OTUs to a larger number of genera and estimating higher relative abundances for these rare taxa [7]. These differences subsequently impacted beta diversity measurements between samples.
A separate evaluation using human fecal samples confirmed these trends, noting that "the use of different bioinformatic pipelines affects the estimation of the relative abundance of gut microbial community, indicating that studies using different pipelines cannot be directly compared" [11]. The study observed statistically significant differences in relative abundance estimates for all phyla and most abundant genera across pipelines [11].
Table 2: Performance Comparison Based on Empirical Studies
| Performance Metric | mothur | QIIME/QIIME 2 | Notes |
|---|---|---|---|
| Sensitivity for Rare Taxa | Higher (more genera identified) | Lower | Particularly with GreenGenes database [7] |
| Richness Estimation | Higher (P < 0.05) | Lower | More favorable rarefaction curves [7] |
| Analytical Specificity | Lower for rare taxa | Higher for rare taxa | mothur may overestimate rare taxa [7] |
| Database Dependence | Significant | Significant | SILVA reduces inter-pipeline differences [7] |
| Reproducibility Across OS | Minimal differences [11] | Minimal differences [11] | Both show good cross-platform consistency |
Reference Database Considerations
The performance differences between pipelines are modulated by the choice of reference database. The SILVA database has been shown to attenuate discrepancies between mothur and QIIME, producing more comparable richness, diversity, and relative abundance estimates for common rumen microbes [7] [15]. This has led to recommendations that "the SILVA database seemed a preferred reference dataset for classifying OTUs from rumen microbiota" [7] when using either pipeline.
Recent QIIME 2 developments have expanded database support, including the incorporation of updated Greengenes2 classifiers [12] and enhanced functionality for creating custom reference databases through plugins like RESCRIPt [13]. The platform's modular architecture facilitates accommodation of new reference datasets as they become available.
Experimental Protocols and Workflows
Standardized Analytical Workflows
The following protocols outline core analytical pathways for both ecosystems, representing standardized approaches for 16S rRNA amplicon analysis:
Detailed Methodological Considerations
mothur Protocol follows a sequential processing approach where quality filtering, alignment to reference databases (SILVA recommended), and distance-based clustering (typically at 97% similarity) form the core workflow [7]. The pipeline produces Operational Taxonomic Units (OTUs) through heuristic algorithms that bin sequences based on similarity thresholds. Critical parameters include quality score thresholds, alignment method (e.g., NAST-based aligners), and clustering algorithm selection (e.g., average neighbor) [16].
QIIME 2 Protocol employs a more modular approach, with denoising algorithms (DADA2 or Deblur) that model and correct sequencing errors to resolve Amplicon Sequence Variants (ASVs) [17] [16]. This method provides single-nucleotide resolution without predefined clustering thresholds. The platform's plugin system allows specialized tools to be incorporated at each step, with provenance tracking automatically recording all parameters and software versions [12].
Multi-Amicon Sequencing Application
For comprehensive taxonomic profiling, multi-amplicon sequencing approaches targeting multiple variable regions have been developed. A recently validated QIIME 2 and R-based pipeline for semiconductor-based sequencing demonstrates the platform's adaptability to complex experimental designs [18]. This standardized workflow integrates data from all targeted 16S regions, generating microbial profiles comparable to proprietary software while maintaining the advantages of open-source transparency and reproducibility [18].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents and Resources for 16S rRNA Analysis
| Resource | Function | Pipeline Compatibility |
|---|---|---|
| SILVA Database | Curated 16S/18S rRNA reference database for taxonomic classification | Both (recommended for consistency) [7] |
| GreenGenes Database | 16S rRNA gene database and taxonomy reference | Both [7] |
| DADA2 Algorithm | Error correction and ASV inference for single-nucleotide resolution | QIIME 2 (as plugin) [17] |
| UNITE Database | Fungal ITS reference database for taxonomic assignment | QIIME 2 (via q2-feature-classifier) [12] |
| RESCRIPt Plugin | Reference database curation and management for custom markers | QIIME 2 [13] |
| Mock Community Standards | Validation and benchmarking of pipeline performance | Both (essential for quality control) [16] |
The QIIME and mothur ecosystems have fundamentally shaped the landscape of 16S rRNA analysis through their complementary philosophical approaches. mothur offers a streamlined, efficient solution with predictable performance characteristics, while QIIME 2 provides an extensible framework with robust provenance tracking and a rapidly evolving method repertoire.
Current evidence suggests that pipeline selection meaningfully impacts analytical outcomes, particularly for low-abundance taxa and beta diversity assessments [7] [11] [17]. The field is increasingly moving toward ASV-based approaches (as implemented in QIIME 2) for improved resolution and cross-study comparability [17] [16], though OTU-based methods retain value for specific applications and historical comparisons.
As the microbiome research field matures, standardization and reproducibility become increasingly critical. The development of validated, open-source pipelines like the QIIME 2-based multi-amplicon workflow [18] represents an important step toward mitigating technical variability and enhancing biological discovery. Both platforms continue to evolve, with recent updates focusing on improved visualization, enhanced database support, and more sophisticated analytical capabilities [14] [12].
Researchers should select their analytical pipeline based on specific experimental requirements, computational resources, and the need for methodological comparability with existing datasets. Whichever ecosystem is chosen, consistent application throughout a study, transparent reporting of parameters, and validation using mock communities remain essential practices for generating robust, interpretable results in microbiome research.
In 16S rRNA gene amplicon sequencing, raw sequence data is processed into structured units that enable quantitative microbial community analysis. The field has primarily utilized two types of units: Operational Taxonomic Units (OTUs) and Amplicon Sequence Variants (ASVs) [19]. OTUs are clusters of sequences grouped based on a predefined similarity threshold, traditionally 97%, which is intended to approximate the species level [20] [21]. In contrast, ASVs are unique sequences inferred from the data through a process of error correction and resolution of single-nucleotide differences, providing a higher-resolution, reproducible representation of microbial diversity without relying on arbitrary clustering thresholds [20] [19] [22]. The final output from both methods is a feature table—either an OTU or ASV table—which is a matrix detailing the abundance of each unit in every sample, forming the basis for all subsequent ecological and statistical analyses [23] [22].
Table 1: Core Concepts: OTUs vs. ASVs
| Feature | Operational Taxonomic Units (OTUs) | Amplicon Sequence Variants (ASVs) |
|---|---|---|
| Definition | Clusters of sequences with a defined similarity (e.g., 97%) [21] | Exact, error-corrected sequences differing by as little as one nucleotide [20] [23] |
| Basis | Identity clustering based on a fixed dissimilarity threshold [19] | Denoising based on statistical error models and probability [20] [19] |
| Typical Resolution | Approximate (e.g., species-level with 97% identity) [20] | High (single-nucleotide) [22] |
| Reproducibility | Variable, depends on clustering method and parameters [19] | High, results are consistent across studies [19] [22] |
| Primary Method | Clustering (de novo, closed-reference, open-reference) [21] [19] | Denoising (e.g., DADA2, Deblur) [20] [22] |
Quantitative Comparisons and Pipeline Performance
The choice of bioinformatics pipeline and reference database can significantly impact the resulting microbial community composition. A study comparing two widely used pipelines, QIIME and mothur, on rumen microbiota from dairy cows revealed both consistencies and critical differences [7] [8].
Table 2: Pipeline and Database Comparison: Mothur vs. QIIME
| Aspect | Finding | Implication |
|---|---|---|
| Abundant Genera (RA > 1%) | High agreement between mothur and QIIME on identity and relative abundance, regardless of database (GreenGenes or SILVA) [7] [8] | Core, high-abundance community members are robustly identified across pipelines. |
| Less Abundant Genera (RA < 10%) | Significant differences (P < 0.05) with GreenGenes; mothur assigned OTUs to more genera and at larger relative abundances [7] [8] | Low-abundance and rare biosphere are more sensitive to analytical choices. |
| Taxonomical Richness | Mothur consistently clustered sequences into a larger number of OTUs, resulting in higher observed richness [7] [8] | mothur may exhibit higher analytical sensitivity, particularly for rare taxa. |
| Database Choice | Differences were attenuated, but not erased, when using the SILVA database instead of GreenGenes [7] [8] | SILVA is preferred for rumen microbiota, leading to more comparable results between pipelines. |
Furthermore, comparisons between OTU and ASV methods show that while the overall biological conclusions about community differences (beta diversity) can be robust, the taxonomic profiles are most comparable at higher taxonomic levels (e.g., family) or when using a 99% OTU identity threshold coupled with frequency filters to remove low-abundance clusters [20].
Experimental Protocols for 16S rRNA Data Analysis
Protocol 1: Constructing an ASV Table with DADA2
The following protocol outlines the steps for constructing a feature table using the DADA2 denoising algorithm within the QIIME 2 environment [20] [22].
Input: Paired-end FASTQ files (demultiplexed, primers removed). Software: QIIME 2 (incorporating DADA2), R [20] [22]. Database: SILVA or GreenGenes for taxonomic assignment [7] [21].
Data Preprocessing and Quality Control:
- Quality Assessment: Use
FastQCto visualize sequence quality profiles [21]. - Trimming and Filtering: Based on quality profiles, trim sequences to remove low-quality bases and filter out sequences with ambiguous bases or expected errors. For the V3-V4 region, forward reads are often trimmed to 280bp and reverse reads to 240bp, or as dictated by the quality plot [22].
- Quality Assessment: Use
Core Denoising with DADA2:
- Learn Error Rates: The algorithm learns the specific error rates of the sequencing run from the data itself, creating an error model [22].
- Dereplication and Denoising: Combine identical sequences and apply the core sample inference algorithm to distinguish true biological sequences from sequencing errors [19] [22].
- Merge Paired-end Reads: Merge the denoised forward and reverse reads, requiring a minimum overlap (e.g., 12bp) and removing mismatches [20].
- Remove Chimeras: Identify and remove chimeric sequences using the
removeBimeraDenovofunction [22].
Construct ASV Table: The output of DADA2 is a sequence table (matrix) reporting the frequency of each non-chimeric, denoised ASV in every sample [22].
Taxonomic Assignment: Assign taxonomy to each ASV by comparing sequences to a reference database using a classifier (e.g., the Naive Bayes classifier in QIIME2) [21].
Protocol 2: Generating OTUs via Open-Reference Clustering
This protocol describes a hybrid method for OTU picking that leverages reference databases while retaining novel sequences, often implemented in QIIME or mothur [21] [19].
Input: High-quality, preprocessed sequences (e.g., stitched, filtered). Software: QIIME or mothur. Database: GreenGenes or SILVA for reference clustering [7] [21].
Initial Closed-Reference Clustering:
- Compare all quality-filtered sequences against a reference database.
- Sequences that match a reference sequence at or above a defined identity threshold (e.g., 97%) are assigned to that reference-based OTU [19].
De Novo Clustering of Failures:
OTU Table Generation:
- The results from the closed-reference and de novo clustering steps are combined into a single OTU table.
- A representative sequence is selected for each OTU (e.g., the most abundant sequence) [21].
Taxonomic Assignment and Chimera Removal:
- Assign taxonomy to the representative sequences using a classifier and reference database.
- Perform chimera detection and removal (e.g., with UCHIME) on the representative sequences or the initial dataset [21].
Table 3: Key Resources for 16S rRNA Gene Analysis
| Category | Item | Function and Application |
|---|---|---|
| Bioinformatics Software | QIIME/QIIME 2 [7] [22] | A comprehensive, modular pipeline for processing amplicon data from raw sequences to statistical analysis. |
| mothur [7] [8] | A single-piece, standalone software pipeline for analyzing 16S rRNA gene sequences. | |
| DADA2 [20] [22] | An R package that performs denoising to infer ASVs from amplicon data with high resolution. | |
| Reference Databases | SILVA [7] [21] | A curated database of aligned rRNA sequences; often preferred for non-human microbiomes like rumen [7]. |
| GreenGenes [7] [21] | A reference database for bacterial and archaeal 16S rRNA gene sequences, historically widely used. | |
| Experimental Controls | Mock Community [23] | A defined mix of microbial cells or DNA with known composition; used as a positive control to evaluate sequencing and bioinformatics performance. |
| No-Template Control (NTC) [23] | A water blank carried through DNA extraction and library preparation to identify laboratory and reagent contamination. | |
| Sequencing Platforms | Illumina MiSeq/HiSeq [22] | High-throughput platforms for short-read sequencing, commonly used for 16S amplicon studies (e.g., V4 region). |
In the field of microbiome research, the choice of sequencing method is a fundamental decision that shapes the scope, resolution, and cost of a study. Two primary techniques dominate this landscape: amplicon sequencing (typically targeting the 16S rRNA gene for bacteria and archaea) and shotgun metagenomic sequencing. Within the context of developing a bioinformatics pipeline for 16S rRNA data analysis, understanding the capabilities and limitations of each method is paramount. Amplicon sequencing, analyzed through pipelines like QIIME and mothur, provides a cost-effective means of taxonomic profiling, whereas shotgun sequencing offers a more comprehensive view of the entire microbial community, including its functional potential [24] [25]. This application note delineates the core concepts of these techniques, provides a structured comparison, and offers clear guidelines for selecting the appropriate method, supported by detailed experimental protocols and data from key studies.
Core Technological Principles
16S rRNA Amplicon Sequencing
The 16S ribosomal RNA (rRNA) gene is a cornerstone of microbial phylogeny and taxonomy. This gene contains nine hypervariable regions (V1-V9), which are flanked by conserved regions. The principle of 16S amplicon sequencing involves using polymerase chain reaction (PCR) with primers designed to bind to these conserved regions, thereby amplifying the intervening hypervariable regions [25] [26]. The resulting PCR amplicons are then sequenced using high-throughput platforms. The hypervariable sequences serve as unique fingerprints, allowing for the identification and classification of bacteria and archaea present in a sample.
The bioinformatics analysis of these sequences, using pipelines such as QIIME and mothur, involves several standardized steps [27] [28]. These include quality filtering, merging of paired-end reads, removal of chimeric sequences, and clustering of sequences into Operational Taxonomic Units (OTUs) or resolving Amplicon Sequence Variants (ASVs). These units are then classified taxonomically by comparing them to reference databases like SILVA or Greengenes [29] [28].
Shotgun Metagenomic Sequencing
In contrast to the targeted approach of amplicon sequencing, shotgun metagenomic sequencing is an untargeted method that fragments all DNA in a sample— microbial, host, and otherwise— into countless small pieces [24] [25]. These fragments are sequenced in a high-throughput manner, generating millions of short reads. Bioinformatics pipelines then assemble these reads into longer contigs or directly align them to comprehensive genomic databases. This process allows for the simultaneous identification of microorganisms across all domains of life (bacteria, archaea, viruses, fungi, and protists) and enables the reconstruction of metabolic pathways and the annotation of gene functions [25] [30].
The following diagram illustrates the fundamental procedural differences between these two sequencing strategies.
The choice between 16S amplicon and shotgun sequencing involves trade-offs between cost, resolution, and analytical depth. The table below synthesizes quantitative and qualitative data from recent studies to provide a clear, side-by-side comparison.
Table 1: Comprehensive comparison of 16S rRNA amplicon sequencing and shotgun metagenomic sequencing.
| Factor | 16S rRNA Amplicon Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Typical Cost (USD per sample) | ~$50 [24] | Starting at ~$150 (depends on depth) [24] |
| Principle | Targeted PCR amplification of a specific gene region [25] | Untargeted, random sequencing of all DNA [25] |
| Taxonomic Resolution | Genus-level (sometimes species) [24] [30] | Species-level and often strain-level [24] [30] |
| Taxonomic Coverage | Bacteria and Archaea only [24] | All domains: Bacteria, Archaea, Viruses, Fungi [24] [30] |
| Functional Profiling | No (only predicted, e.g., with PICRUSt) [24] | Yes (direct identification of genes and pathways) [24] [30] |
| Bioinformatics Complexity | Beginner to Intermediate [24] | Intermediate to Advanced [24] |
| Sensitivity to Host DNA | Low (PCR targets microbes) [24] | High (can be mitigated by sequencing depth) [24] |
| Reference Databases | Well-established (e.g., SILVA, Greengenes) [29] | Growing, but complex (e.g., NCBI, GTDB, UHGG) [29] |
| Data Sparsity & Diversity | Sparser data, lower alpha diversity [29] | More complete community profile, higher alpha diversity [29] |
| Species-Level Detection | Detects only part of the community [29] [31] | Reveals a wider diversity, including low-abundance species [29] [31] |
Experimental Protocols and Benchmarking Data
To ground this comparison in empirical evidence, we review the methodologies and key findings from two pivotal studies that directly compared these sequencing techniques.
Protocol 1: Comparative Analysis in Colorectal Cancer Microbiota
- Study Objective: To compare the reliability of 16S and shotgun sequencing for bacterial profiling in the context of colorectal cancer (CRC), advanced lesions, and healthy controls [29].
- Sample Collection: 156 human stool samples were collected from a CRC screening program. Samples were stored at -20°C by participants and then preserved at -80°C upon delivery [29].
- DNA Extraction: Two different kits were used for compatibility with each sequencing method: the NucleoSpin Soil Kit (for shotgun) and the Dneasy PowerLyzer Powersoil kit (for 16S) [29].
- Sequencing:
- 16S rRNA: The V3-V4 hypervariable region was amplified and sequenced. An in-house bioinformatics pipeline using DADA2 and additional classification with BLASTN and Kraken2 against the SILVA database was used to enhance species-level classification [29].
- Shotgun: Whole genome sequencing was performed, and human reads were filtered out using Bowtie2 against the GRCh38 human genome [29].
- Key Findings: The study concluded that 16S sequencing detects only a portion of the gut microbiota community revealed by shotgun sequencing. Shotgun data was less sparse and exhibited higher alpha diversity. While machine learning models from both techniques could identify microbial signatures associated with CRC (e.g., Parvimonas micra), only some shotgun models showed predictive power in an independent test set. The study recommends shotgun sequencing for in-depth analysis of stool samples, while noting 16S remains suitable for studies with more targeted aims [29].
Protocol 2: Taxonomic Characterization in Chicken Gut Microbiota
- Study Objective: To evaluate the agreement between 16S and shotgun sequencing for taxonomic characterization of the chicken gut microbiota under different experimental conditions [31].
- Experimental Design: The same DNA samples from a previous intervention study were re-analyzed using both 16S and shotgun sequencing. Conditions included different gastrointestinal tract compartments (crop and caeca) and sampling times [31].
- Methodology: The study focused on comparing relative species abundance distributions, rarefaction curves, and the power of each method to discriminate between experimental conditions using differential abundance analysis (DESeq2) [31].
- Key Findings: Shotgun sequencing, when a sufficient number of reads was available (>500,000), identified a statistically significant higher number of taxa, primarily the less abundant ones. The genera detected only by shotgun sequencing were biologically meaningful and able to discriminate between experimental conditions as effectively as the more abundant genera detected by both methods. The correlation of genus abundances between the two techniques was strong (average Pearson's r = 0.69) for shared taxa, but 16S failed to detect many of the significant changes identified by shotgun sequencing [31].
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table catalogues critical reagents and materials required for executing the sequencing protocols described in the cited studies.
Table 2: Key Research Reagent Solutions for 16S and Shotgun Sequencing Workflows.
| Item | Function/Application | Example Product(s) / Methods |
|---|---|---|
| DNA Extraction Kit | Isolation of high-quality, inhibitor-free microbial DNA from complex samples. | PowerSoil DNA Isolation Kit [32], NucleoSpin Soil Kit [29], Dneasy PowerLyzer Powersoil kit [29] |
| 16S PCR Primers | Amplification of specific hypervariable regions of the 16S rRNA gene. | Primers for V3-V4 region [29] [26], V1-V3 region [26] |
| Library Prep Kit | Preparation of DNA fragments for sequencing, including end-repair, adapter ligation, and index PCR. | NEBNext Ultra DNA Library Prep Kit [32], NEXTflex 16S V1–V3 Amplicon-Seq kit [32] |
| Bioinformatics Pipelines | Software for processing raw sequencing data, taxonomic assignment, and diversity analysis. | QIIME [24] [27], mothur [24] [28], DADA2 [29] |
| Reference Databases | Curated collections of genomic or gene sequences for taxonomic and functional classification. | 16S: SILVA [29], Greengenes [33]. Shotgun: NCBI RefSeq [29], GTDB [29] |
| Computational Resources | Hardware and software environment for data-intensive bioinformatics analysis. | Galaxy platform [28], High-performance computing (HPC) clusters |
Decision Framework: When to Use 16S rRNA Sequencing
The following workflow diagram encapsulates the decision-making process for selecting the appropriate sequencing method, based on research goals, sample type, and resources.
Guidance for Pipeline Development
For researchers building a bioinformatics pipeline for 16S rRNA data analysis using tools like QIIME and mothur, it is critical to recognize the inherent limitations of the data these tools process.
- Taxonomic Resolution: The pipeline will typically achieve reliable genus-level classifications, but species-level identification is limited and dependent on the hypervariable region chosen [33]. Full-length 16S sequencing on long-read platforms can improve this but is not yet the standard.
- Functional Insights: Since 16S data does not directly provide functional information, pipelines often integrate prediction tools like PICRUSt [24] [28]. These predictions are inferences based on reference genomes and should be interpreted with caution.
- Bias Awareness: The pipeline must account for biases introduced during PCR amplification and the choice of primers, which can affect the observed taxonomic composition [29].
Both 16S amplicon and shotgun metagenomic sequencing provide powerful yet distinct lenses for examining microbial communities. 16S rRNA sequencing remains a highly cost-effective and accessible method for large-scale studies focused primarily on the taxonomy of bacteria and archaea, making it an excellent tool for initial surveys and hypothesis generation. In contrast, shotgun metagenomic sequencing offers superior taxonomic resolution, broader kingdom coverage, and direct access to functional insights, at a higher cost and computational burden. The decision between them is not a matter of which is universally better, but which is the most appropriate tool for the specific research question, sample type, and available resources. As the field advances, the development of robust, standardized bioinformatics pipelines for both methods, particularly within user-friendly platforms like Galaxy, is essential for ensuring the reproducibility and translational impact of microbiome research [28].
In the standardized bioinformatics pipeline for 16S rRNA data analysis using tools like QIIME and Mothur, the initial computational processing represents merely the final stage of a long analytical chain. Preceding this, the critical wet-lab decision of primer selection irrevocably shapes all downstream results. Targeted amplicon sequencing of the 16S ribosomal RNA gene serves as the cornerstone method for profiling microbial communities across diverse environments, from the human gut to engineered bioreactors [34] [35]. The 16S rRNA gene contains nine hypervariable regions (V1-V9) flanked by conserved sequences that enable primer design for PCR amplification [36]. However, no universal primer pair exists that perfectly amplifies all bacterial taxa without bias, making primer choice a fundamental determinant of observed microbial composition [37] [38].
The growing recognition of primer-induced biases challenges the assumption that data generated using different hypervariable regions are directly comparable. Recent comprehensive studies demonstrate that specific taxa are systematically underrepresented or completely missing from taxonomic profiles when using suboptimal primer combinations [34] [39]. For instance, the Bacteroidetes phylum may be missed using primers 515F-944R (targeting V4-V5), while the V1-V2 region fails to adequately capture Fusobacteriota without specific modifications [34] [39]. These technical artifacts can lead to biologically erroneous conclusions if not properly accounted for in experimental design.
This Application Note examines how hypervariable region selection biases 16S rRNA sequencing results, provides structured comparisons of primer performance characteristics, and offers practical protocols for optimizing primer choice within standardized bioinformatics pipelines for microbial ecology research.
Theoretical Foundation: Mechanisms of Primer Bias
Primer binding efficiency varies substantially across bacterial taxa due to several molecular mechanisms. Sequence mismatch tolerance differs among DNA polymerases, leading to preferential amplification of templates with perfect primer complementarity [38]. The degeneracy design of primers (incorporating mixed bases at highly variable positions) attempts to mitigate this but introduces variability in primer synthesis efficiency and annealing kinetics [38]. Additionally, secondary structure formation in template regions can block primer access, particularly in GC-rich sequences [38].
Perhaps the most underappreciated source of bias stems from intergenomic variation within conserved regions. Traditionally, primer design has assumed that flanking regions remain sufficiently conserved across all bacteria for universal amplification. However, comprehensive analysis of 20 core gut genera reveals substantial variability even in these supposedly conserved regions [37]. Shannon entropy analysis demonstrates unexpected nucleotide variation at primer binding sites, challenging the concept of truly "universal" primers and explaining systematic amplification failures for specific bacterial lineages [37].
Variable Region Performance Characteristics
Different hypervariable regions offer varying levels of taxonomic resolution due to their distinct evolutionary rates and sequence characteristics. The V4 region provides reliable family-level classification but struggles with species-level discrimination for many taxa, while the V1-V2 regions often enable finer taxonomic resolution but may miss certain phyla [40] [39]. The V3-V4 region, popularized by the Earth Microbiome Project, offers a compromise between length and discriminative power but exhibits particularly problematic off-target amplification in human-derived samples [39] [41].
Table 1: Taxonomic Resolution of Commonly Used Hypervariable Regions
| Hypervariable Region | Optimal Taxonomic Level | Notable Limitations | Recommended Applications |
|---|---|---|---|
| V1-V2 | Genus to species | Poor coverage of Fusobacteriota without modifications | Human biopsy samples, clinical diagnostics |
| V3-V4 | Family to genus | High off-target amplification of human DNA | Environmental samples, stool samples |
| V4 | Family | Limited species-level resolution | Broad microbial surveys, educational use |
| V4-V5 | Family | May miss Bacteroidetes | Industrial microbiome monitoring |
| V6-V8 | Genus | Variable coverage across Firmicutes | Specialized environmental applications |
Experimental Evidence: Documented Impacts of Primer Choice
Comparative Performance Across Sample Types
The severity of primer-induced bias varies considerably across sample types, largely dependent on the ratio of host to bacterial DNA. In human biopsy samples where host DNA predominates (often exceeding 97% of total DNA), primer pairs targeting the V3-V4 region generate 70-98% human-derived sequences in gastrointestinal tract biopsies, breast tissue, and esophageal samples [39] [41]. This massive off-target amplification drastically reduces sequencing depth for bacterial communities and can obscure rare taxa. Switching to V1-V2 targeted primers reduces human DNA alignment to nearly zero while significantly increasing observable bacterial richness [39].
In contrast, stool samples with minimal human DNA contamination show much smaller differences between primer sets, though taxonomic composition shifts remain substantial [34] [35]. Similarly, mock communities with known composition reveal that certain primer pairs consistently fail to detect specific members, with the magnitude of bias increasing with community complexity [34] [36].
Quantitative Comparison of Primer Performance
Systematic evaluation of 57 commonly used primer sets against the SILVA database identified three candidate primers (V3P3, V3P7, and V4_P10) that provide balanced coverage across 20 key genera of the core gut microbiome [37]. Notably, many widely used "universal" primers showed significant limitations in coverage, failing to amplify substantial portions of microbial diversity due to unexpected variability in conserved regions [37].
Table 2: Performance Metrics of Select Primer Pairs in Human Gut Microbiome Profiling
| Primer Pair | Target Region | Average Coverage (%) | Human DNA Amplification | Taxonomic Richness (ASVs) | Reproducibility (R²) |
|---|---|---|---|---|---|
| 68F-338R (V1-V2M) | V1-V2 | 89.7 | <0.1% | 215 ± 34 | 0.96 |
| 341F-785R | V3-V4 | 85.3 | 34-98% | 127 ± 42 | 0.87 |
| 515F-806R | V4 | 82.1 | 55-85% | 98 ± 39 | 0.83 |
| 515F-944R | V4-V5 | 79.4 | <0.1% | 142 ± 28 | 0.91 |
| 1115F-1492R | V7-V9 | 76.8 | <0.1% | 135 ± 31 | 0.89 |
Practical Protocols: A Methodological Framework
Protocol 1: In Silico Primer Validation
Purpose: Computational assessment of primer performance against reference databases before wet-lab experimentation.
Materials:
- Reference database (SILVA, GreenGenes, or RDP)
- Primer sequences in FASTA format
- Computational tools: TestPrime, DECIPHER, or mopo16S
Procedure:
- Database Selection: Download and curate an appropriate 16S rRNA reference database (e.g., SILVA SSU Ref NR 99%).
- Primer Screening: Use TestPrime or equivalent tool to calculate coverage statistics for your primer candidates against the database.
- Mismatch Analysis: Identify potential systematic mismatches, particularly at the 3'-end of primers where they most impact amplification.
- Taxonomic Specificity: Evaluate coverage variation across phyla of interest, noting any systematic exclusions.
- Amplicon Length Verification: Confirm that the resulting amplicon size is compatible with your sequencing platform.
Interpretation: Primer pairs achieving ≥70% coverage across all target phyla and ≥90% coverage for at least four out of 20 representative genera are considered candidates for experimental validation [37].
Protocol 2: Experimental Validation with Mock Communities
Purpose: Empirical evaluation of primer performance using synthetic microbial communities of known composition.
Materials:
- Mock community with staggered or even abundance (e.g., ZymoBIOMICS Gut Microbiome Standard)
- DNA extraction kit suitable for your sample type
- Selected primer pairs for comparison
- High-fidelity PCR master mix
- Access to appropriate sequencing platform
Procedure:
- Sample Preparation: Extract DNA from mock community according to manufacturer's protocol, including negative controls.
- Library Preparation: Amplify using candidate primer pairs with identical cycling conditions and PCR cycle numbers.
- Sequencing: Process all samples in the same sequencing run to minimize technical variation.
- Bioinformatic Processing: Process raw data through standardized pipeline (QIIME2 or Mothur) with identical parameters.
- Bias Quantification: Compare observed composition to expected composition using Bray-Curtis dissimilarity or similar metrics.
Interpretation: Successful primer pairs should recover all expected taxa with relative abundances correlating strongly (R² > 0.85) with expected composition [34] [36].
Protocol 3: Optimization for Host-Associated Microbiomes
Purpose: Specialized protocol for samples with high host DNA content (biopsies, blood, etc.).
Materials:
- V1-V2 targeted primers (68F-338R with modifications)
- High-fidelity PCR enzyme with GC-rich buffer capability
- Human DNA depletion kit (optional)
- Agarose gel electrophoresis equipment
Procedure:
- Human DNA Depletion: Optional step using selective hybridization or enzymatic digestion to reduce host DNA.
- Primer Modification: For V1-V2 primers, include modified forward primer (68F_M) to capture Fusobacteriota.
- Annealing Optimization: Implement touchdown PCR with annealing temperatures between 62-65°C to enhance specificity.
- Cycle Number Titration: Use minimum PCR cycles needed for sufficient library yield (typically 25-30 cycles).
- Size Selection: Purify amplicons of correct size (≈260bp for V1-V2) to exclude primer dimers and non-specific products.
Interpretation: Successful implementation yields >90% bacterial reads with representative diversity across known community members [39].
Bioinformatics Considerations: Pipeline Adjustments for Primer Effects
Database Selection and Customization
The reference database used for taxonomic assignment must align with the amplified region to avoid misclassification. Region-specific training sets significantly improve classification accuracy in QIIME2 and Mothur [34]. For example, using a V1-V2 trained classifier with V4 sequence data introduces substantial misclassification errors. Database nomenclature differences (e.g., Enterorhabdus versus Adlercreutzia) further complicate cross-study comparisons using different primer sets [34].
Truncation Length Optimization
Different hypervariable regions require specific quality trimming parameters to maximize data quality. Systematic testing reveals that appropriate truncation of amplicons is essential, and different truncated-length combinations should be empirically determined for each primer set and study [34]. For the commonly used V3-V4 region (341F-785R), truncation at 260bp forward and 200bp reverse typically optimizes quality without excessive data loss, while V1-V2 amplicons (68F-338R) perform best with 220bp forward and 180bp reverse truncation [39].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Primer Evaluation and Optimization
| Reagent/Resource | Specifications | Application | Example Sources |
|---|---|---|---|
| Mock Community B | 20 bacterial strains, even and staggered configurations | Protocol validation | BEI Resources, ATCC |
| ZymoBIOMICS Gut Standard | 19 bacterial and archaeal strains with multiple 16S copies | Primer bias assessment | Zymo Research |
| SILVA SSU Database | Curated 16S rRNA sequences with quality checking | In silico validation | silva-arb.org |
| TestPrime Tool | Online primer coverage analysis | Primer screening | silva-arb.org |
| mopo16S Software | Multi-objective primer optimization | Novel primer design | http://sysbiobig.dei.unipd.it |
| High-Fidelity Polymerase | Low error rate, minimal bias | Library preparation | Multiple vendors |
Decision Framework: Visual Guide to Primer Selection
The following workflow diagram provides a systematic approach to primer selection based on experimental goals and sample characteristics:
Primer selection constitutes a fundamental, often underestimated source of bias in 16S rRNA sequencing studies that propagates through all downstream bioinformatics analyses. The evidence presented demonstrates that hypervariable region choice systematically impacts observed microbial composition, diversity metrics, and taxonomic resolution. To maximize experimental validity:
- Validate primers empirically using mock communities that reflect your sample type's complexity
- Select variable regions based on your specific sample characteristics and research questions
- Account for primer effects when comparing datasets generated with different amplification protocols
- Document primer sequences and amplification conditions thoroughly to enable appropriate cross-study comparisons
No single hypervariable region provides optimal resolution for all research scenarios. Rather, researchers should align primer selection with specific experimental goals while acknowledging the technical constraints inherent in targeted amplicon sequencing. When studying novel environments or when primer biases may substantially impact conclusions, employing a multi-primer approach or supplementing with PCR-independent methods (such as metatranscriptomics or shotgun sequencing) provides valuable verification of community composition [42]. Through deliberate primer selection and appropriate validation, researchers can minimize technical artifacts and focus on meaningful biological variation within their microbial systems.
Step-by-Step Workflows: From Sequence Data to Community Analysis
Within the broader context of bioinformatics pipelines for 16S rRNA data analysis, the mothur tool suite represents a cornerstone methodology for processing amplicon sequencing data. Developed by the Schloss lab, this SOP provides a robust framework for analyzing microbial communities using sequences generated from Illumina's MiSeq platform [43] [44]. The protocol outlined here exemplifies the application of this pipeline to investigate a specific biological question: understanding the effect of normal variation in the gut microbiome on host health, using a longitudinal study of mouse fecal samples [43] [45]. This SOP has been extensively validated through peer-reviewed research and continues to be refined as the field advances [43].
The primary strength of the mothur pipeline lies in its comprehensive approach to error reduction and data curation. Unlike approaches that may sacrifice data quality for throughput, this methodology employs rigorous quality control measures that reduce error rates by as much as two orders of magnitude, providing a reliable foundation for downstream ecological interpretations [44]. The protocol processes paired-end reads that overlap in the V4 region of the 16S rRNA gene (approximately 253 bp), leveraging Illumina's sequencing technology to generate high-quality data for microbial community analysis [43].
Experimental Design and Workflow
The mothur MiSeq SOP follows a logical progression from raw sequencing data to interpreted ecological patterns, with multiple quality checkpoints throughout the process. Figure 1 illustrates the complete workflow from initial setup through final analysis.
Figure 1. Overview of the mothur MiSeq SOP workflow. The diagram illustrates the sequential steps from raw sequencing data processing through alignment, quality control, and final diversity analysis. Reference databases are incorporated at critical junctures to ensure proper sequence alignment and taxonomic classification.
Computational Requirements and Setup
The mothur pipeline can be implemented across various computing environments, from personal computers to high-performance computing clusters. The software is written in C++, is independent of operating system, and requires no dependencies [46]. For larger datasets (e.g., >100 samples), computational resources should be scaled accordingly, with recommended specifications including sufficient memory (≥100 GB RAM for large projects) and multiple processors to leverage mothur's parallel computing capabilities [28] [46].
The initial setup involves creating a dedicated project directory and obtaining the necessary reference files:
- Create project directory: Organize a dedicated folder structure for the analysis
- Obtain reference files: Download the SILVA-based bacterial reference alignment, RDP training set, or alternative databases [43]
- Install mothur: Obtain the latest version of mothur executable [43]
Materials and Reagents
Research Reagent Solutions
Table 1: Essential computational reagents and reference databases for the mothur MiSeq SOP
| Item | Function | Source |
|---|---|---|
| mothur Software | Primary analysis platform for processing 16S rRNA sequences | mothur.org [43] |
| SILVA Reference Database | Reference alignment for sequence alignment and classification | SILVA [43] |
| RDP Training Set | Reference taxonomy for Bayesian classification | RDP [43] |
| Greengenes Database | Alternative reference database for classification | Greengenes [7] |
| Illumina MiSeq Sequences | Raw paired-end FASTQ files from 16S rRNA amplicon sequencing | Experimental data [43] |
| Mock Community DNA | Control sample with known composition for error rate assessment | e.g., HMP_MOCK.v35.fasta [43] |
Sample Data Collection
The exemplary dataset referenced in this SOP was generated from a longitudinal study investigating gut microbiome dynamics in mice. Fresh feces were collected from mice on a daily basis for 365 days post-weaning, focusing specifically on comparing the rapid change period (first 10 days) with a stable period (days 140-150) [43]. To make the tutorial tractable, a subset of this data is provided, representing one animal at 10 time points (5 early and 5 late) plus a mock community sample [43]. The mock community, composed of genomic DNA from 21 bacterial strains, provides an essential control for measuring pipeline error rates and their effect on downstream analyses [43].
Step-by-Step Protocol
Data Preparation and Contig Assembly
The initial phase of the pipeline focuses on organizing input data and assembling paired-end reads into contigs:
Create files list: Execute the
make.filecommand to identify forward and reverse reads and create a stability.files document that maps sample names to their respective FASTQ files [43]:Assemble contigs: Use the
make.contigscommand to combine paired-end reads, creating the reverse complement of the reverse read and joining reads into contigs [43]:This command employs a quality-aware algorithm that resolves disagreements between paired reads by considering quality scores, requiring a quality score difference of ≥6 points when both sequences have a base, or a score >25 when one sequence has a base and the other has a gap [43].
Sequence summary: Generate initial quality metrics using
summary.seqsto assess sequence length distribution and quality [46]:
Quality Control and Alignment
This critical phase removes low-quality sequences and aligns reads to a reference database:
Screen sequences: Remove sequences with ambiguous bases, excessive length, or homopolymers
Align to reference: Align screened sequences to the appropriate reference database (e.g., SILVA) [28]:
Filter alignment: Remove poorly aligned regions and minimize overhangs
Remove redundant sequences: Dereplicate to reduce computational burden
Pre-cluster sequences: Implement a lightweight clustering to reduce sequencing errors
Chimera Removal and Classification
This phase identifies and removes artificial chimeric sequences while assigning taxonomy:
Chimera detection: Identify chimeras using UCHIME or ChimeraSlayer [28]:
Remove chimeras: Filter identified chimeras from the dataset
Taxonomic classification: Assign taxonomy using a Bayesian classifier with an appropriate reference training set [28]:
OTU Clustering and Diversity Analysis
The final phase generates operational taxonomic units and calculates diversity metrics:
Calculate distances: Generate a distance matrix for clustering
Cluster sequences: Cluster sequences into OTUs using an appropriate algorithm (e.g., Opticlust, average neighbor) at 97% similarity [28] [16]:
Classify OTUs: Assign consensus taxonomy to each OTU
Calculate diversity metrics: Generate alpha and beta diversity measures, including rarefaction curves and ordination plots [45]:
Expected Results and Interpretation
Sequence Processing Statistics
When properly executed, the pipeline should yield high-quality data with minimal errors. The mock community sample provides a critical benchmark for assessing pipeline performance.
Table 2: Expected sequence processing statistics at major pipeline stages
| Processing Stage | Expected Result | Quality Indicator |
|---|---|---|
| Initial Contig Assembly | >70% of read pairs successfully assembled | Sequencing quality and library preparation |
| Post-Quality Control | <5% of sequences removed during screening | Initial sequence quality |
| Chimera Removal | 10-30% of sequences identified as chimeric | PCR amplification artifacts |
| Final OTU Clustering | Error rate <0.1% on mock community | Overall pipeline accuracy [44] |
| Taxonomic Classification | >90% of sequences classified to genus level | Reference database appropriateness |
Mock Community Analysis
The mock community with known composition serves as a vital control for quantifying error rates. When processing the 21-strain mock community, the pipeline should correctly identify the expected sequences with minimal errors. Studies have demonstrated that this SOP can reduce error rates by as much as two orders of magnitude compared to uncorrected data [44]. The error rate can be calculated by comparing the observed sequences to the expected reference sequences in the mock community.
Diversity Assessment
For the exemplary mouse gut microbiome data, the pipeline should reveal differences in community structure between early (days 0-10) and late (days 140-150) time points. Alpha diversity metrics (e.g., Shannon index, Chao1) may show higher variability during the early rapid change period compared to the stable late period. Beta diversity measures (e.g., Weighted Unifrac) should demonstrate clustering of samples by time period, indicating distinct community structures.
Troubleshooting and Technical Notes
Common Issues and Solutions
- Low sequence yield after make.contigs: Adjust
trimoverlapparameter and check primer sequences in oligos file [46] - Poor alignment rates: Verify that the reference database matches the target region (e.g., V4 region for V4 primers)
- High chimera rates: Optimize PCR cycle numbers in wet-lab protocol and consider using multiple chimera detection methods
- Low classification rates: Try alternative reference databases (SILVA, Greengenes, RDP) as performance varies by sample type [43]
Methodological Considerations
The choice between OTU clustering and Amplicon Sequence Variants (ASVs) represents a key methodological decision. While this SOP focuses on traditional OTU clustering at 97% similarity, ASV approaches (e.g., DADA2, Deblur) offer single-nucleotide resolution and may reduce spurious OTUs [16]. Recent benchmarking analyses indicate that OTU algorithms (like those in mothur) tend to achieve clusters with lower errors but with more over-merging, while ASV algorithms produce more consistent output but may suffer from over-splitting [16].
When analyzing data from multiple studies with different primer sets or sequencing regions, it is generally recommended to analyze datasets separately rather than attempting to combine them in a single analysis, as alignment artifacts can lead to significant data loss [47].
Alternative Implementations
For researchers seeking a more user-friendly interface, the Galaxy mothur Toolset (GmT) provides a web-based implementation of the entire mothur tool suite, making the pipeline accessible to non-bioinformaticians while maintaining analytical rigor [28]. This implementation preserves all functionality while adding workflow automation and integration with visualization tools like Krona and Phinch [28] [45].
Comparative studies have shown that mothur and QIIME produce comparable results for abundant taxa (>10% relative abundance) but may differ in their handling of rare taxa, with mothur typically assigning OTUs to a larger number of genera for less abundant microorganisms [7]. The choice between these platforms may depend on the specific research question and the importance of detecting rare community members.
Within the broader context of developing robust bioinformatics pipelines for 16S rRNA data analysis, the transition from traditional OTU-clustering methods to Amplicon Sequence Variants (ASVs) represents a significant advance. ASVs offer higher resolution by inferring exact biological sequences, thereby reducing the spurious inflation of diversity metrics common with arbitrary OTU clustering [48]. QIIME 2 has emerged as a comprehensive, reproducible framework that integrates these modern denoising methods, notably through its DADA2 and Deblur plugins, into a cohesive analysis workflow [49] [50]. This protocol details the application of these plugins within the QIIME 2 environment, providing a standardized pipeline for researchers and drug development professionals to process raw 16S rRNA sequencing data into high-resolution, analytically powerful ASVs.
Background: Denoising vs. Clustering
The initial step in marker gene analysis involves grouping similar sequences. This can be achieved through two primary approaches: denoising and clustering [51].
- Denoising (ASV Workflow): Methods like DADA2 and Deblur correct sequence errors and remove chimeras to infer the exact biological sequences present in the original sample, resulting in Amplicon Sequence Variants (ASVs) [48] [52]. This approach provides higher resolution and is often more sensitive than OTU clustering.
- Clustering (OTU Workflow): Methods like VSEARCH group sequences based on a percent similarity threshold (e.g., 97%), creating Operational Taxonomic Units (OTUs). This was the standard approach before denoising methods became prevalent [51].
For most applications, denoising is recommended as it provides a more accurate representation of biological sequences without relying on arbitrary similarity thresholds [52] [51]. The following workflow focuses on this modern ASV-based approach.
The complete QIIME 2 workflow for ASV inference, from raw data to biological insight, involves multiple stages that can be visualized in the following diagram. The denoising step, which is the focus of this protocol, is central to this process.
Detailed Experimental Protocol
Initial Data Preparation and Import
Principle: All data used in QIIME 2 must be imported into a QIIME 2 artifact (.qza file) to ensure type safety and provenance tracking [52] [51]. For raw sequencing data, the most straightforward approach uses a manifest file.
Protocol:
- Create a manifest file: This is a tab-separated text file that maps sample identifiers to the file paths of their forward and reverse (for paired-end) reads [48] [52]. The format must be exact: sample-id | forward-absolute-filepath | reverse-absolute-filepath F26 | /path/to/F261.fq.gz | /path/to/F262.fq.gz F27 | /path/to/F271.fq.gz | /path/to/F272.fq.gz
Import sequences: Use the
qiime tools importcommand, specifying the type of data and the format. For paired-end data with Phred33 quality scores, the command is [48]:Summarize and visualize imported data: Generate an interactive quality plot to determine optimal truncation parameters for denoising [48]:
Open the resulting
.qzvfile at https://view.qiime2.org to inspect quality score distributions and read lengths.
Denoising with DADA2
Principle: DADA2 models and corrects Illumina amplicon errors to infer exact amplicon sequence variants (ASVs), performing quality filtering, dereplication, chimera removal, and read merging (for paired-end data) in a single process [48] [53].
Protocol:
- Execute DADA2 denoising: Based on the quality plots, choose truncation lengths (
--p-trunc-len-fand--p-trunc-len-r) where median quality scores drop significantly (e.g., below Q30). The reads must still overlap after truncation [48] [53].
Key Parameters:
--p-trim-left: Number of nucleotides to trim from the 5' start of reads to remove primers or low-quality bases.--p-trunc-len: Position to truncate reads at the 3' end due to quality drop. Reads shorter than this are discarded.--p-max-ee: Maximum expected errors allowed in a read; reads with higher expected errors are discarded.--p-n-threads: Number of threads to use for parallel processing to speed up computation on multi-core systems [53].
Summarize outputs: Generate visualizations to inspect the feature table, representative sequences, and denoising statistics [48].
Denoising with Deblur
Principle: Deblur uses an error-profile-based approach to remove sequencing errors from Illumina data, resulting in ASVs. It is typically applied to single-end reads and can include a positive filter step for 16S data [52].
Protocol:
- Join paired-end reads (if necessary): Deblur operates on single-end reads. If you have paired-end data and wish to use Deblur, join reads first using
q2-vsearch[51].
- Execute Deblur denoising: For 16S data, use the
denoise-16Saction, which performs a positive filtering step against reference sequences. For other markers (e.g., ITS), usedenoise-other[52]. The--p-trim-lengthparameter is required for Deblur to ensure all reads are the same length for analysis.
Critical Decision Points: DADA2 vs. Deblur
The choice between DADA2 and Deblur depends on your data type and analytical goals. The table below summarizes the key differences to guide your selection.
Table 1: Comparison of DADA2 and Deblur for ASV Inference in QIIME 2
| Feature | DADA2 | Deblur |
|---|---|---|
| Primary Use Case | Paired-end or single-end Illumina reads [53] [52] | Primarily single-end Illumina reads [52] |
| Read Joining | Performs read merging internally as part of denoise-paired [52] |
Requires pre-joined reads via a separate tool (e.g., vsearch join-pairs) [51] |
| Algorithm Core | Error model based on alternation of nucleotides and quality scores [48] | Error profile based on read shifts and specific substitutions [52] |
| Positive Filter (16S) | No positive filtering; classifies all input reads | Optional positive filter against reference database in denoise-16S [52] |
| Key Parameter | --p-trunc-len-f/-r (Truncation length) [53] |
--p-trim-length (Trim all reads to fixed length) [52] |
| Output | Feature table, representative sequences, denoising stats [48] [53] | Feature table, representative sequences, deblurring stats [52] |
Downstream Analysis
Following ASV inference, the resulting feature table and representative sequences form the basis for all subsequent biological interpretations.
Taxonomic Classification: Assign taxonomy to ASVs using a pre-trained classifier [48].
Diversity Analysis: Calculate alpha and beta diversity metrics, which often require building a phylogenetic tree [49] [51].
Visualization: Create interactive barplots and other visualizations to explore taxonomic composition and diversity results [48].
The Scientist's Toolkit
A successful QIIME 2 analysis requires several key components, from raw data to reference databases. The following table details these essential resources.
Table 2: Essential Research Reagents and Resources for QIIME 2 ASV Analysis
| Item | Specifications & Function | Example Sources |
|---|---|---|
| Raw Sequence Data | Demultiplexed FASTQ files (Phred33 encoding). The starting point of the analysis. | Illumina MiSeq/HiSeq instruments; BaseSpace [54] |
| Sample Metadata File | Tab-separated values (.tsv) file with sample-id column and experimental factors. Links biological samples to their data and covariates. |
Researcher-generated; validated with Keemei [55] |
| QIIME 2 Environment | Installed and activated Conda environment. Provides the core platform and all integrated plugins for analysis. | https://docs.qiime2.org [50] [56] |
| Taxonomic Classifier | Pre-trained Naive Bayes classifier artifact (.qza). Used for assigning taxonomy to ASV sequences. | SILVA, Greengenes; or custom-trained with fit-classifier-naive-bayes [48] |
| Reference Databases | Curated sequences and taxonomy files (e.g., FASTA, .txt). Used for classifier training or, in Deblur, for positive filtering. | SILVA, Greengenes, GTDB, UNITE [57] |
| Quality Visualization Tool | Web-based interface for viewing .qzv files. Essential for interactive quality control and result exploration. |
https://view.qiime2.org [48] [50] |
Integrating DADA2 or Deblur within the QIIME 2 framework provides a powerful, standardized, and reproducible pipeline for inferring high-resolution ASVs from 16S rRNA sequencing data. This protocol outlines the critical steps and decision points, empowering researchers to move beyond traditional OTU clustering. The structured workflow from raw data import through denoising to downstream analysis ensures robust, transparent, and reproducible results, forming a solid bioinformatics foundation for microbiome studies in both basic research and drug development contexts.
Within bioinformatics pipelines for 16S rRNA data analysis, such as those implemented in QIIME and mothur, the pre-processing of raw sequencing data is a critical foundational step. The accuracy of all downstream ecological inferences—including taxonomic assignment, diversity analysis, and statistical comparison—is fundamentally dependent on the rigorous application of quality filtering, paired-end read merging, and chimera removal [58] [59]. This protocol outlines detailed methodologies for these key pre-processing steps, providing a standardized framework that ensures data quality and reproducibility in microbial ecology studies. The procedures are designed to be applicable within popular analysis environments, including QIIME, mothur, and USEARCH, and are essential for researchers, scientists, and drug development professionals working with 16S amplicon sequencing data.
The Scientist's Toolkit: Essential Research Reagents & Software
The following table catalogues key software tools and reference databases essential for implementing a robust 16S rRNA pre-processing pipeline.
Table 1: Key Research Reagent Solutions for 16S rRNA Data Pre-processing
| Item Name | Type | Primary Function in Pre-processing |
|---|---|---|
| USEARCH / VSEARCH [60] [59] | Software Tool | Paired-end read merging, quality filtering, dereplication, and chimera checking. VSEARCH is an open-source alternative to USEARCH. |
| DADA2 [58] | Software Tool | A denoising algorithm that infers amplicon sequence variants (ASVs) by modeling and correcting Illumina-sequenced amplicon errors. |
| mothur [45] [59] | Software Pipeline | A comprehensive, open-source software package for processing 16S rRNA gene sequences, including all steps from raw data to statistical analysis. |
| PEAR [61] | Software Tool | An ultrafast, memory-efficient, and highly accurate paired-end read merger. |
| Chimera Slayer [62] | Software Algorithm | A tool for detecting chimeric sequences by identifying reads that are hybrids of multiple parent sequences. |
| SILVA Database [58] [63] | Reference Database | A curated, high-quality alignment of ribosomal RNA genes used for sequence alignment, chimera checking, and taxonomic assignment. |
| GreenGenes Database [58] [63] | Reference Database | A curated 16S rRNA gene database used for taxonomic classification and phylogenetic analysis. |
The pre-processing of 16S rRNA amplicon data follows a logical sequence to transform raw sequencing reads into a high-quality set of non-chimeric sequences ready for downstream analysis. The following diagram illustrates the core workflow and the key decision points at each stage.
The execution of this workflow relies on established quantitative thresholds to ensure consistency and data integrity. The following table summarizes the key parameters and their typical values for Illumina MiSeq data targeting the V3-V4 hypervariable region.
Table 2: Key Quantitative Parameters for 16S rRNA Pre-processing Steps
| Processing Step | Key Parameter | Typical Value / Range | Rationale & Reference |
|---|---|---|---|
| Quality Filtering | Phred Quality Score | ≥ 20 [64] | Removes bases with a base call accuracy of < 99%. |
| Minimum Read Length | > 100 bp [64] | Discards uninformatively short fragments. | |
| Expected Errors (maxee) | ≤ 1.0 [60] | Filters reads with an unacceptably high number of expected errors. | |
| Read Merging | Minimum Overlap | 10-20 bp [61] | Ensures sufficient sequence for reliable alignment. |
| Merged Length (V3-V4) | 440 - 470 bp [60] | Filters reads that are too long or short, indicating poor merges or off-target amplification. | |
| Chimera Removal | Parent Divergence | Detectable from ~4% [62] | Chimera Slayer is sensitive to chimeras from parents with low sequence divergence. |
Detailed Experimental Protocols
Protocol 1: Initial Quality Assessment and Paired-End Read Merging
This initial protocol assesses the raw data quality and merges the forward and reverse reads to reconstruct the full-length amplicon.
Methodology:
- Overall Quality Check with FastQC: Pool all forward and reverse reads from a sequencing run into separate files and run FastQC to generate an overview of read quality, per-base sequence quality, and potential adapter contamination [60].
- Merge Paired-End Reads: Use a merging tool to align the overlapping regions of forward and reverse reads. This can be done with or without strict quality filtering at this stage to retain as many reads as possible for downstream mapping [60].
- Using USEARCH:
- Using PEAR: PEAR is an alternative, highly accurate merger that evaluates all possible overlaps and uses a statistical test [61].
Protocol 2: Quality Filtering and Primer Removal
Following merging, sequences undergo rigorous quality filtering, and amplicon primer sequences are stripped.
Methodology:
- Quality Filtering: If not applied during merging, perform expected error filtering to remove low-quality sequences. Expected error filtering is superior to simple quality score averaging as it sums the probabilities of errors across the entire read [60].
- Strip Amplicon Primers: Remove the primer sequences used for amplification. This is critical as primers are prone to errors and are not part of the biological sequence of interest. Use a mapping file that specifies the forward and reverse primer sequences for each sample and a custom script to detect and remove them [60].
- Convert to FASTA Format: Convert the final quality-filtered, primer-removed FASTQ file to FASTA format for downstream steps, as quality scores are no longer required.
Protocol 3: Chimera Detection and Removal
Chimeric sequences are PCR artifacts formed from two or more biological parent sequences, leading to false inflation of diversity. Their removal is non-optional.
Methodology:
- Understand Chimera Formation: Chimeras form during PCR when an incomplete extension product from one sequence acts as a primer on a different, related template in a subsequent cycle [62]. Their formation is reproducible and influenced by template abundance and similarity.
- Select a Chimera Detection Tool: Several tools exist, with varying sensitivities.
- Chimera Slayer: A sensitive algorithm that can detect chimeras from parents with as little as 4% divergence. It works by comparing each query sequence to a database of non-chimeric reference sequences, identifying potential parents, and flagging sequences that are more closely related to an in-silico chimera of the parents than to any real sequence [62]. The following diagram details its logical operation.
- Execute Chimera Checking: Run the chosen chimera detection tool on the entire set of quality-filtered sequences. This step is typically integrated into larger pipelines like mothur and QIIME.
- Within mothur: The
chimera.vsearchcommand is commonly used to identify and remove chimeric sequences against a reference database like SILVA. - Best Practice: Always use a well-curated, high-quality reference database (e.g., SILVA, GreenGenes) specific to your chimera checker for optimal results [62] [45].
- Within mothur: The
Concluding Remarks
The pre-processing protocols detailed herein—encompassing stringent quality control, accurate read merging, and sensitive chimera removal—constitute the essential first chapter in any robust bioinformatics thesis on 16S rRNA analysis. Adherence to these standardized methods ensures that the resulting feature table (whether OTUs or ASVs) is a reliable representation of the underlying microbial community, providing a solid foundation for all subsequent ecological and statistical interpretations. As the field progresses, the core principles of quality filtering and artifact removal will remain paramount, regardless of advancements in sequencing technologies or analytical algorithms.
The analysis of microbial communities through 16S rRNA gene amplicon sequencing is a cornerstone of modern microbial ecology and microbiome research. The bioinformatic processing of this sequencing data has undergone a significant methodological evolution, shifting from traditional Operational Taxonomic Unit (OTU) clustering to the more recent Amplicon Sequence Variant (ASV) approach [65] [66]. This shift is central to pipelines such as QIIME and mothur, which represent two of the most widely used bioinformatics suites in this field. The choice between OTU clustering and ASV denoising is not merely technical but has profound implications on the resolution, reproducibility, and biological interpretation of microbial diversity data [67] [68]. This application note details the core differences between these methods, their performance characteristics, and provides structured experimental protocols for their implementation within a comprehensive 16S rRNA analysis pipeline.
Theoretical Foundation: OTUs vs. ASVs
Operational Taxonomic Units (OTUs)
OTUs are clusters of sequencing reads grouped based on a user-defined sequence similarity threshold, typically 97%, which historically was intended to approximate bacterial species-level differentiation [65] [66]. This method reduces dataset complexity and computational load by grouping similar sequences, which can help mitigate the impact of sequencing errors as erroneous reads are merged with correct biological sequences during clustering [68].
- Advantages: OTU clustering is somewhat tolerant of sequencing errors, computationally efficient for large datasets, and facilitates comparison with a vast body of historical research [65] [69].
- Disadvantages: The method loses fine-scale resolution by grouping sequences, uses an arbitrary similarity threshold that may not reflect true biological boundaries and can inflate alpha-diversity measures by generating spurious clusters [67] [65].
Amplicon Sequence Variants (ASVs)
ASVs represent unique, error-corrected biological sequences distinguished by single-nucleotide differences without relying on arbitrary clustering thresholds [65] [68]. Denoising algorithms, such as those in DADA2 and USEARCH-UNOISE3, employ statistical models to differentiate true biological variation from sequencing noise, resulting in a set of high-resolution sequence variants.
- Advantages: ASVs offer single-nucleotide resolution, enabling the detection of closely related microbial strains. They are highly reproducible across studies because they represent exact sequences, provide more accurate taxonomic assignment, and effectively correct for sequencing errors [67] [65] [69].
- Disadvantages: ASV generation is computationally intensive and may over-split biological variants if not properly tuned, though modern algorithms are designed to mitigate this [65].
The following workflow illustrates the fundamental procedural differences between the OTU-clustering and ASV-denoising approaches in a typical 16S rRNA analysis pipeline.
Performance Comparison and Benchmarking Data
The choice between OTU and ASV methodologies significantly impacts the outcome of microbial community analysis. Benchmarking studies using mock communities (samples with known composition) and diverse environmental samples have quantified these differences in terms of sensitivity, specificity, and effects on diversity metrics.
Table 1: Performance Comparison of Common Bioinformatics Pipelines on a Mock Microbial Community [67]
| Pipeline | Type | Sensitivity | Specificity | Key Findings |
|---|---|---|---|---|
| DADA2 | ASV | Best | Lower | Highest sensitivity to detect true variants, but at the expense of slightly lower specificity. |
| USEARCH-UNOISE3 | ASV | High | Best Balance | Offers the best balance between resolution (sensitivity) and specificity. |
| Qiime2-Deblur | ASV | High | High | Strong performance, comparable to UNOISE3. |
| USEARCH-UPARSE | OTU | Moderate | Lower | Good performance for an OTU-based method, but lower specificity than ASV pipelines. |
| MOTHUR | OTU | Moderate | Lower | Performs well, but with lower specificity than ASV-level pipelines. |
| QIIME-uclust | OTU | Low | Lowest | Produces a large number of spurious OTUs and inflates alpha-diversity; not recommended. |
Table 2: Impact of Pipeline Choice on Ecological Diversity Metrics [66] [68]
| Diversity Metric | Impact of OTU vs. ASV Choice | Notes |
|---|---|---|
| Richness (Alpha) | Strong Effect | OTU methods (especially QIIME-uclust) often overestimate richness compared to ASV methods [67]. The discrepancy can be attenuated by rarefaction [66]. |
| Beta Diversity | Strong Effect | The choice affects presence/absence indices (e.g., unweighted Unifrac) more than abundance-weighted indices (e.g., weighted Unifrac) [66]. |
| Taxonomic Composition | Significant Discrepancies | Identification of major classes and genera shows significant discrepancies, especially for low-abundance taxa [66] [7]. |
| Rare Taxa Detection | Variable | OTU clustering may retain more rare sequences, but with a higher risk of being spurious. DADA2 (ASV) is highly sensitive for low-abundance sequences [69]. |
The following chart synthesizes data from benchmark studies to illustrate the relative performance of different pipelines in detecting true biological signals while controlling errors.
Experimental Protocols
Protocol 1: OTU Clustering with Mothur
This protocol follows the standard operating procedure (SOP) for MiSeq data in mothur, clustering sequences into OTUs at a 97% identity threshold [68].
1. Sample Processing and Sequencing:
- DNA Extraction: Use a standardized kit (e.g., PowerSoil Pro Kit) on your sample material (fecal, soil, tissue, etc.).
- Library Preparation: Amplify the V4 region of the 16S rRNA gene using primers 515F/806R with attached Illumina adapters and barcodes.
- Sequencing: Perform 2x250 bp paired-end sequencing on an Illumina MiSeq platform.
2. Bioinformatics Analysis with Mothur:
- Software: Mothur (v1.8.0 or later).
- Reference Databases: SILVA database for alignment and taxonomic classification.
- Steps:
- Merge Paired-End Reads:
make.contigs() - Screen Sequences:
screen.seqs()to remove sequences of unusual length or with ambiguous bases. - Alignment:
align.seqs()using the SILVA reference alignment. - Filter Alignment:
filter.seqs()to remove poorly aligned regions. - Pre-clustering:
pre.cluster()to slightly denoise by merging very similar sequences. - Chimera Removal:
chimera.vsearch()with default parameters. - OTU Clustering:
cluster.split()ordist.seqs()followed bycluster()at 97% identity. - Taxonomic Classification:
classify.seqs()using the Wang method and the SILVA taxonomy. - OTU Table Generation:
make.shared()to create the final OTU table.
- Merge Paired-End Reads:
Protocol 2: ASV Inference with DADA2
This protocol utilizes the DADA2 algorithm within the QIIME2 framework or R environment for superior resolution [67] [68].
1. Sample Processing and Sequencing: (Identical to Protocol 1)
2. Bioinformatics Analysis with DADA2:
- Software: DADA2 (v1.7.0 or later) in R or via the QIIME2 plugin (q2-dada2).
- Reference Databases: SILVA or GreenGenes for taxonomic assignment post-denoising.
- Steps:
- Quality Profiling: Inspect read quality plots using
plotQualityProfile()in R. - Filter and Trim:
filterAndTrim()to truncate reads based on quality scores and remove low-quality reads. - Learn Error Rates:
learnErrors()to create an error model specific to your dataset. - Dereplication:
derepFastq()to combine identical reads. - Core Denoising:
dada()to infer sample composition and correct errors. - Merge Paired Reads:
mergePairs()to create the full-length denoised sequences. - Construct Sequence Table:
makeSequenceTable()to build the ASV table. - Remove Chimeras:
removeBimeraDenovo(). - Taxonomic Assignment:
assignTaxonomy()against a reference database.
- Quality Profiling: Inspect read quality plots using
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for 16S rRNA Amplicon Sequencing Workflows
| Item | Function / Application | Example Product / Specification |
|---|---|---|
| DNA Extraction Kit | Isolation of high-quality microbial genomic DNA from complex samples. | PowerSoil Pro Kit (Qiagen) [68], Quick-DNA Fecal/Soil Microbe Miniprep Kit (Zymo Research) [20]. |
| PCR Master Mix | Robust amplification of the 16S rRNA target region. | Five Prime Hot Master Mix [67]. |
| 16S rRNA Primers | Target-specific amplification of hypervariable regions. | 515F/806R for V4 region [67] [68]; 338F/533R for V3 region [20]. |
| Sequencing Standard | Validating sequencing run performance and bioinformatic pipeline accuracy. | ZymoBIOMICS Microbial Community Standard [69]; BEI Resources Mock Community [67]. |
| Size Selection Beads | Purification of amplified libraries to remove primer dimers and short fragments. | AMPure XP beads (Beckman Coulter) [67] [20]. |
| Sequencing Platform | Generation of paired-end amplicon sequences. | Illumina MiSeq with V2/V3 reagent kits (2x250 bp) [67] [7]. |
| Reference Database | Taxonomic classification of OTUs or ASVs. | SILVA (release 132 or later) [7] [68], GreenGenes (13_5 or later) [7] [20]. |
The field of 16S rRNA amplicon analysis is experiencing a definitive shift from OTU-based clustering to ASV-based denoising methods due to the latter's superior resolution, reproducibility, and accuracy [67] [65] [69]. Based on the synthesized evidence:
- For new studies seeking high-resolution insights, ASV-based pipelines (DADA2, USEARCH-UNOISE3, Qiime2-Deblur) are strongly recommended. They are particularly advantageous for detecting strain-level variation and for cross-study comparisons.
- USEARCH-UNOISE3 offers an excellent balance of sensitivity and specificity, while DADA2 provides the highest sensitivity, making it suitable for detecting low-abundance taxa [67] [69].
- OTU-based methods (e.g., Mothur) remain valid for comparing new data with legacy datasets generated using the same methodology. However, the use of QIIME-uclust is discouraged due to its propensity to generate spurious OTUs [67].
- The choice of pipeline has a greater impact on alpha and beta diversity measures than other common methodological choices like rarefaction or OTU identity threshold (97% vs. 99%) [66] [68].
- For taxonomic classification, the SILVA database is often preferred, as it can help attenuate differences between pipelines compared to GreenGenes [7] [8].
In conclusion, the selection of a bioinformatic pipeline should be a deliberate decision aligned with the study's goals. While ASV methods represent the current standard for accuracy, understanding the properties and limitations of both OTU and ASV approaches is essential for robust 16S rRNA data analysis and interpretation.
Within bioinformatics pipelines for 16S rRNA data analysis, such as QIIME and mothur, taxonomic classification is a foundational step that translates genetic sequence data into biological insights about microbial community composition. The choice of reference database is critical, as it directly influences the taxonomic labels assigned, the diversity metrics calculated, and, consequently, all subsequent ecological interpretations and hypotheses [70]. Among the most widely used databases are SILVA and Greengenes, each with distinct histories, curation philosophies, and performance characteristics. This application note provides a detailed comparison of these databases, evaluates their impact on taxonomic classification, and outlines structured protocols to guide researchers in selecting and implementing the appropriate resource within their bioinformatics pipelines.
SILVA Database
The SILVA database (from Latin silva, forest) is a comprehensive resource for quality-checked and aligned ribosomal RNA sequence data for all three domains of life (Bacteria, Archaea, and Eukarya) [71] [72]. Its taxonomy is heavily curated, integrating information from authoritative resources including Bergey's Manual of Systematic Bacteriology, the List of Prokaryotic Names with Standing in Nomenclature (LPSN), and, more recently, the Genome Taxonomy Database (GTDB) for higher taxonomic ranks [72] [73]. A key feature of SILVA is its reliance on phylogenies inferred from small subunit rRNAs, with manual curation playing a significant role in the process [74] [73]. The database is regularly updated, with releases such as 138.2 (July 2024) providing large, comprehensive datasets (SSU Parc with over 9 million sequences) as well as refined, high-quality subsets (SSU Ref NR 99) suitable for reference-based classification [71].
Greengenes Database
The original Greengenes database was dedicated to Bacteria and Archaea and distinguished itself by providing chimera screening and standard alignments [75]. However, its development stalled after the May 2013 release (gg135_99), limiting its coverage of newly discovered taxa [70] [76]. A significant recent development is the introduction of Greengenes2 in 2024, a complete redesign from the ground up [77]. This new version is backed by whole genomes and integrates the GTDB taxonomy with the Living Tree Project, aiming to harmonize analysis between 16S rRNA and shotgun metagenomic datasets [77]. Unlike the original, Greengenes2 is constructed from a phylogenomic tree based on hundreds of marker genes, onto which millions of 16S rRNA sequences are placed [77].
Table 1: Core Characteristics of SILVA and Greengenes Databases
| Feature | SILVA | Greengenes (Legacy) | Greengenes2 (2024.09) |
|---|---|---|---|
| Domains Covered | Bacteria, Archaea, Eukarya [72] | Bacteria & Archaea [75] | Bacteria & Archaea [77] |
| Primary Curational Basis | SSU rRNA phylogeny & manual curation [73] | De novo tree from 16S, rank mapping from NCBI [74] | Phylogenomic tree (Web of Life), GTDB taxonomy integration [77] |
| Update Status | Regular releases (e.g., 138.2 in 2024) [71] | Static since 2013 [70] [76] | Actively developed (2024.09) [77] |
| Key Strength | Comprehensive, manually curated, covers Eukaryotes | Historical default in QIIME; chimera-checked [75] | Modern, genome-backed, harmonizes 16S & shotgun data [77] |
| Notable Taxonomy Sources | Bergey's, LPSN, GTDB, UniEuk [73] | NCBI, Bergey's (via RDP) [75] [74] | GTDB, Living Tree Project [77] |
Table 2: Quantitative Database Comparison from Mock Community Analysis [70]
| Performance Metric | SILVA | Greengenes (Legacy) | EzBioCloud |
|---|---|---|---|
| True Positive Genera (approx.) | ~35 | ~30 | >40 |
| False Positive Genera | Highest (~20% of predictions) | Moderate | Lowest |
| Species-Level Accuracy | Moderate (decreased vs. genus) | Poor (few correct species) | Good (highest among tested) |
| Richness (Observed OTUs) | Overestimated | Overestimated | Most accurate to expected |
| Evenness (Simpson's Index) | Underestimated | Underestimated | Most accurate to expected |
Experimental Protocol: Database Evaluation with a Mock Community
A robust method for evaluating the accuracy of taxonomic classification databases involves using a mock microbial community, where the exact composition of strains is known beforehand. This allows for a direct comparison between the taxonomic assignments generated by a bioinformatics pipeline and the ground truth.
Materials and Reagents
- Mock Community Genomic DNA: A commercially available standard comprising an even mix of genomic DNA from known bacterial strains (e.g., ZymoBIOMICS Microbial Community Standard).
- 16S rRNA Gene Primers: Primers targeting the appropriate hypervariable region (e.g., V3-V4: 341F/806R).
- Sequencing Kit: Reagents for library preparation and sequencing on an Illumina MiSeq, NovaSeq, or similar platform.
- Bioinformatics Workstation: A computer server with sufficient memory (≥16 GB RAM) and multi-core processors.
- Software Pipelines: QIIME 2 (2024.5 or later) or mothur (v.1.48.0 or later) installed, for example, via Conda environments.
- Reference Databases: Downloaded and formatted reference sequences and taxonomies for SILVA (e.g., release 138.2) and Greengenes2 (2024.09).
Detailed Methodology
1. Sequence Data Generation and Preprocessing:
- Perform 16S rRNA gene amplification and paired-end sequencing (2x300 bp) of the mock community according to manufacturer protocols.
- Import raw sequencing data (in FASTQ format) into your chosen pipeline.
- Perform quality control: in QIIME 2, use q2-demux to visualize quality plots, followed by q2-dada2 for denoising, quality filtering, merging of paired-end reads, and chimera removal. This produces a feature table of amplicon sequence variants (ASVs) and their representative sequences [77].
2. Taxonomic Classification against Multiple Databases:
- Classifier Training: Train a Naïve Bayes classifier on each reference database using the specific primer pair.
- For QIIME 2: Use the feature-classifier fit-classifier-naive-bayes method on the extracted reference reads.
- Classification: Assign taxonomy to the ASVs using the trained classifiers.
- For QIIME 2: Use the classify-sklearn action against each trained classifier to generate separate taxonomy tables for SILVA and Greengenes2.
3. Accuracy Assessment: - Create a ground truth taxonomy table based on the known composition of the mock community. - Genus-Level Analysis: For each database, compare the assigned genera against the ground truth. Calculate: - True Positives (TP): Correctly identified genera. - False Positives (FP): Genera reported that are not in the community. - False Negatives (FN): Genera present in the community but not identified. - Species-Level Analysis: Repeat the above comparison at the species level. - Diversity Indices: Calculate alpha diversity indices (e.g., Observed OTUs, Shannon, Simpson) from the feature table. Compare these calculated values against the known, even distribution of the mock community. An accurate database will yield richness and evenness values closer to the expected values [70].
Workflow Visualization
Practical Application and Protocols
Database Selection Guidelines
The choice between databases involves trade-offs. The following decision tree provides a structured approach for researchers.
Protocol: Classifying 16S rRNA Data in QIIME 2
This protocol outlines the steps for taxonomic classification of V4 amplicon data using both SILVA and Greengenes2 in QIIME 2.
1. Data Import and Preprocessing:
- Import demultiplexed paired-end sequences (manifest.csv) into a QIIME 2 artifact (paired-end-demux.qza).
- Denoise and generate ASVs using DADA2: qiime dada2 denoise-paired ... --p-trunc-len-f 220 --p-trunc-len-r 200 --o-representative-sequences rep-seqs.qza --o-table table.qza.
2. Database-Specific Classification:
- For SILVA:
- Download and import the SILVA reference sequences and taxonomy (e.g., for release 138.2).
- Extract reads matching your primer region: qiime feature-classifier extract-reads ... --p-16s-reader silva-138.2-99-seqs.qza ... --o-reads silva-138.2-99-515f-806r.qza.
- Train a classifier: qiime feature-classifier fit-classifier-naive-bayes ... --i-reference-reads silva-138.2-99-515f-806r.qza --i-reference-taxonomy silva-138.2-99-tax.qza --o-classifier silva-138.2-99-515f-806r-classifier.qza.
- Classify your ASVs: qiime feature-classifier classify-sklearn ... --i-classifier silva-138.2-99-515f-806r-classifier.qza --i-reads rep-seqs.qza --o-classification taxonomy-silva.qza.
- For Greengenes2:
- The process is streamlined via the q2-greengenes2 plugin.
- Download the Greengenes2 taxonomy artifact: wget http://ftp.microbio.me/greengenes_release/current/2024.09.taxonomy.asv.nwk.qza.
- Filter your feature table against Greengenes2 and assign taxonomy directly from the phylogeny:
3. Visualization and Comparison:
- Visualize the classified results: qiime metadata tabulate ... --m-input-file taxonomy-silva.qza ... --o-visualization taxonomy-silva.qzv.
- To compare results from both databases, merge the taxonomy tables and ASV sequences, then use visualization tools like an interactive bar plot.
Table 3: Key Resources for 16S rRNA Database Analysis
| Resource | Function/Description | Example Source/Location |
|---|---|---|
| ZymoBIOMICS Microbial Community Standard | Mock community with known composition for validating pipeline and database accuracy. | Zymo Research |
| SILVA SSU Ref NR 99 Dataset | Curated, non-redundant subset of high-quality aligned sequences for reference-based classification. | https://www.arb-silva.de/ |
| Greengenes2 2024.09 Taxonomy Artifact | The new, genome-backed Greengenes2 database in a format ready for use in QIIME 2. | http://ftp.microbio.me/greengenes_release/current/ |
| QIIME 2 (with q2-greengenes2 plugin) | A powerful, extensible, and community-supported bioinformatics pipeline for microbiome analysis. | https://qiime2.org/ |
| mothur | A comprehensive, all-in-one bioinformatics software package for processing 16S rRNA gene sequence data. | https://mothur.org/ |
Taxonomic classification is not a neutral step in 16S rRNA analysis, and the selection of a reference database profoundly impacts biological conclusions. SILVA offers a comprehensive, manually curated, and regularly updated resource that includes Eukaryotes, making it a robust and versatile choice. The legacy Greengenes database is now largely obsolete due to its outdated taxonomy. However, the newly released Greengenes2 represents a significant advance, with its genome-based phylogeny and strong alignment with modern GTDB taxonomy, making it particularly compelling for new studies aiming to integrate 16S and metagenomic data. Researchers should base their choice on the specific needs of their project, considering factors like taxonomic scope, need for modern nomenclature, and compatibility with existing datasets. Whenever possible, validating findings with a mock community or using multiple databases can provide greater confidence in the resulting taxonomic profiles.
In the analysis of 16S rRNA sequencing data within bioinformatics pipelines like QIIME and mothur, downstream analyses comprising alpha diversity, beta diversity, and statistical visualization are crucial for interpreting microbial ecology. Alpha diversity describes the diversity of microbial taxa within a single sample, while beta diversity quantifies the differences in microbial community composition between samples [78]. This protocol details the methodologies for calculating these metrics and generating publication-quality figures, framed within the broader context of a 16S rRNA data analysis pipeline. We provide a standardized set of procedures using QIIME 2 and mothur, two widely adopted platforms in microbiome research [79] [43].
Theoretical Background
Alpha Diversity
Alpha diversity metrics summarize the structure of an ecological community with respect to its richness (number of taxonomic groups), evenness (distribution of abundances of the groups), or both [58]. These metrics can be phylogenetically naive or incorporate the evolutionary relationships between observed taxa.
Beta Diversity
Beta diversity refers to the diversity between samples, representing a measure of how similar or dissimilar samples are to one another [78]. It is typically represented by a distance matrix derived from metrics that can account for phylogenetic relationships (e.g., Weighted or Unweighted UniFrac) or be based solely on organism abundance (e.g., Bray-Curtis) [78] [58].
Materials and Methods
Research Reagent Solutions
The following table lists essential materials and data files required to execute the diversity analyses described in this protocol.
Table 1: Essential Research Reagents and Computational Materials
| Item Name | Function/Description | Example/Format |
|---|---|---|
| Feature Table | Contains counts of each unique sequence variant (ASV) or operational taxonomic unit (OTU) for all samples. | BIOM file (.biom) or QIIME 2 Artifact (.qza). |
| Phylogenetic Tree | Contains the evolutionary relationships between the features (ASVs/OTUs) in the feature table. | Newick file (.tre) or QIIME 2 Artifact (.qza). |
| Sample Metadata | Tab-separated file linking sample IDs to phenotypic and experimental data. | TSV file (.tsv) [80]. |
| Reference Databases | Used for taxonomic assignment and phylogenetic tree construction. | Greengenes, SILVA, or HOMD [43] [58]. |
The downstream analysis workflow begins with a processed feature table and an associated phylogenetic tree. The subsequent steps for calculating and visualizing alpha and beta diversity are illustrated below.
Detailed Experimental Protocol
Calculating Alpha Diversity
Alpha diversity metrics are computed from the feature table. The following commands demonstrate the process in QIIME 2 for both phylogenetic and non-phylogenetic metrics.
QIIME 2 Commands:
- Phylogenetic Metric (Faith's Phylogenetic Diversity): This command calculates Faith PD, which sums the branch lengths of the phylogenetic tree for all taxa present in a sample [81].
- Non-Phylogenetic Metric (Observed OTUs/ASVs): This command calculates the simplest measure of richness: the number of distinct features (e.g., OTUs or ASVs) observed in each sample [81].
A comprehensive list of available alpha diversity metrics is provided in the table below.
Table 2: Selected Alpha Diversity Metrics Available in QIIME 2 [81]
| Metric Name | Type | Description | Key Reference |
|---|---|---|---|
| Faith's PD | Phylogenetic | Sum of the branch lengths of the phylogenetic tree for all taxa in a sample. | Faith (1992) |
| Observed OTUs/ASVs | Non-Phylogenetic | The number of distinct features (OTUs or ASVs) observed in a sample. | DeSantis et al. (2006) |
| Shannon Index | Non-Phylogenetic | Measures richness and evenness; influenced more by rich species than rare ones. | - |
| Pielou's Evenness | Non-Phylogenetic | Measure of how evenly species abundances are distributed. | Pielou (1975) |
| Chao1 Index | Non-Phylogenetic | Estimates the true species richness, including unobserved species. | Chao (1984) |
Calculating Beta Diversity
Beta diversity analysis produces a distance matrix that compares the microbial composition of every sample pair. The following commands are used in QIIME 2.
QIIME 2 Command:
This command calculates the Weighted UniFrac distance, which accounts for the abundance of OTUs/ASVs and their phylogenetic relatedness [78]. For a non-phylogenetic metric, the beta command can be used with, for example, --p-metric braycurtis.
Statistical Analysis of Diversity
Alpha Diversity: To test for significant differences in alpha diversity between groups (e.g., control vs. treatment), parametric (e.g., t-test) or non-parametric (e.g., Kruskal-Wallis) tests can be applied to the alpha diversity vectors. It is critical to have biological replicates within groups to perform statistical testing [82]. If you have more than two groups, an ANOVA can be used [82].
Beta Diversity: The statistical significance of group clustering observed in a beta diversity analysis is typically assessed using a permutation-based non-parametric test like PERMANOVA (Adonis), which is often implemented within QIIME 2's diversity plugins.
Generating Visualizations
Creating intuitive visualizations is a critical final step.
Alpha Diversity Visualization: Alpha diversity data is best visualized using box plots or violin plots, which show the distribution of diversity values within each sample group. These plots can be generated in R using the
phyloseqpackage [58] or from QIIME 2's diversity plugins.Beta Diversity Visualization: The distance matrix generated from beta diversity is visualized using Principal Coordinates Analysis (PCoA) plots. QIIME 2 Command for PCoA:
The resulting ordination can be plotted in Emperor [78]. For publications, 2D plots are often preferred for their clarity over 3D representations [78]. A script like
make_2d_plots.pycan be used for this conversion in QIIME 1, and similar functionality exists in other environments.
Troubleshooting and Technical Notes
- Negative Eigenvalues in PCoA: It is common to encounter small negative eigenvalues when running PCoA. If these values are very small compared to the largest positive eigenvalue, they can be safely ignored, as the transformation to plot them has a negligible impact on the interpretation [78].
- Raw vs. Rarefied Data: There is ongoing debate regarding the need to rarefy (subsample) sequence data before calculating diversity. Some studies demonstrate that weighted UniFrac on raw counts is accurate [78]. Procrustes analysis can be used to compare the results from raw and rarefied data to ensure your conclusions are robust to the normalization method [78].
- Reporting: Researchers must clearly report their analysis protocols, including software versions, diversity metrics used, and any deviations from default parameters, to ensure reproducibility and appropriate comparison between studies [83].
Optimizing Pipeline Performance and Overcoming Common Pitfalls
In the analysis of 16S rRNA gene sequencing data using bioinformatics pipelines such as QIIME and mothur, the selection of critical parameters—specifically, clustering thresholds and quality scores—profoundly impacts the accuracy, resolution, and biological relevance of the resulting microbial community profiles. These choices influence downstream analyses, including taxonomic assignment, alpha- and beta-diversity metrics, and comparative statistics. The established practice of using a 97% similarity threshold for clustering sequences into Operational Taxonomic Units (OTUs) requires re-evaluation in light of modern sequencing technologies and expanded reference databases [84] [33]. Similarly, setting appropriate quality score thresholds is essential for filtering erroneous sequences while retaining biological diversity. This protocol details the critical parameter choices for 16S rRNA data analysis within the QIIME and mothur frameworks, providing evidence-based recommendations and step-by-step methodologies to ensure robust and reproducible microbiome research.
Critical Parameters in 16S rRNA Data Analysis
The following table summarizes the key parameters, their typical values, and the biological and technical considerations researchers must account for.
Table 1: Critical Parameter Choices for 16S rRNA Gene Analysis
| Parameter | Historical Standard | Current Recommendations | Biological & Technical Rationale |
|---|---|---|---|
| Clustering Threshold (OTU) | 97% identity [84] | ~99% for full-length 16S; ~100% for V4 region [84]. Region-specific and taxon-dependent [16]. | The 97% threshold was based on limited historical data. Re-evaluation with finished genomes shows higher thresholds better approximate species-level clusters [84]. |
| Sequence Quality Filtering | Q25 (approx. 0.3% error rate) [85] | Default in QIIME's split_libraries.py: min Q=25 [85]. mothur: maxambig=0, maxhomop=8 [46]. |
Removes low-quality bases and reads with ambiguous base calls or excessive homopolymers, which are potential sources of sequencing error. |
| Target Region Selection | Single hypervariable region (e.g., V4) [34] | Full-length 16S is superior; V1-V3 or V3-V5 are reasonable compromises for short-read platforms [33]. | Different variable regions have varying discriminatory power for specific bacterial taxa. Full-length sequencing captures all variable regions for maximum resolution [34] [33]. |
| Clustering Method | OTU-based (97%) | ASVs (DADA2, Deblur) or OTUs with optimized threshold. ASVs reduce spurious OTUs and allow cross-study comparison [34] [86] [16]. | Denoising algorithms model and correct sequencing errors, resolving single-nucleotide differences. OTU clustering bins sequences based on a fixed identity cutoff. |
Workflow and Decision Pathway for Parameter Selection
The following diagram outlines the logical workflow for analyzing 16S rRNA data, highlighting the critical decision points for parameter selection within a standard bioinformatics pipeline.
Diagram 1: Parameter Selection Workflow for 16S rRNA Analysis. This workflow guides the selection of key parameters based on sequencing technology and analytical goals.
Detailed Experimental Protocols
Protocol 1: QIIME Analysis Workflow with Parameter Optimization
This protocol adapts the standard QIIME pipeline to incorporate critical parameter choices for clustering and quality control, based on the analysis of mouse gut microbial communities [85].
Necessary Resources: A functional installation of QIIME or the QIIME VirtualBox.
Procedure:
Demultiplexing and Initial Quality Filtering: Use the
split_libraries.pyscript to assign multiplexed reads to samples and perform initial quality filtering.- Critical Parameters: The default settings in this command include a minimum quality score of Q25 and require no ambiguous base calls. These parameters are effective for removing low-quality sequences [85].
Picking OTUs and Assigning Taxonomy: Execute the comprehensive workflow script
pick_otus_through_otu_table.pyon the demultiplexed sequences (seqs.fna).- Critical Parameters: This workflow performs several steps. For OTU picking, the default similarity threshold is often 97%. However, as established in Table 1, this should be optimized. To use a more stringent 99% threshold, you would modify the command with parameters specific to the clustering algorithm used (e.g.,
-s 0.99forpick_otus.py). Using a closed-reference OTU picking approach against a curated database can also be beneficial for specific study goals [87].
- Critical Parameters: This workflow performs several steps. For OTU picking, the default similarity threshold is often 97%. However, as established in Table 1, this should be optimized. To use a more stringent 99% threshold, you would modify the command with parameters specific to the clustering algorithm used (e.g.,
Downstream Analysis: The output of the previous step is an OTU table, which can be used for subsequent analyses, including aligning representative sequences, building phylogenetic trees, and calculating diversity metrics [85].
Protocol 2: Mothur Analysis Workflow with Parameter Optimization
This protocol outlines key steps in the mothur pipeline for preparing and clustering sequences, detailing critical parameters as applied to MiSeq data of coral-associated bacteria [46].
Necessary Resources: A functional installation of mothur and sequence files in FASTQ format.
Procedure:
Prepare Files and Assemble Contigs: Use
make.contigs()to combine forward and reverse reads, while simultaneously removing primers and filtering based on quality.- Critical Parameters:
trimoverlap=T: Trims the sequence overlap region, crucial for paired-end reads.pdiffs=2: Allows for up to 2 differences in the primer sequence, accounting for potential sequencing errors or degeneracies in the primers.checkorient=t: Checks the orientation of sequences, which can help recover more reads [46].
- Critical Parameters:
Quality Filtering and Sequence Summary: Further filter sequences based on length, ambiguous bases, and homopolymers. Then, use
summary.seqs()to review sequence characteristics.- Critical Parameters: Standard filtering includes
maxambig=0(no ambiguous bases allowed) andmaxhomop=8(maximum homopolymer length of 8) to eliminate potentially erroneous sequences [46].
- Critical Parameters: Standard filtering includes
Clustering Sequences into OTUs: Cluster the high-quality sequences into OTUs. mothur offers multiple algorithms (e.g.,
cluster.splitusing theopticlustorvsearchmethods).
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key computational tools and databases essential for executing the protocols described above.
Table 2: Research Reagent Solutions for 16S rRNA Analysis
| Item | Function/Description | Example Use in Protocol |
|---|---|---|
| QIIME Software Suite | An open-source bioinformatics pipeline for performing microbiome analysis from raw DNA sequencing data. | Used in Protocol 1 for demultiplexing, OTU picking, and diversity analysis [85]. |
| mothur Software Suite | An open-source, community-developed software that seeks to develop a single method for analyzing community sequencing data. | Used in Protocol 2 for sequence processing, contig assembly, and clustering [46]. |
| SILVA Reference Database | A comprehensive, curated database of aligned ribosomal RNA (rRNA) gene sequences. | Used as a reference for sequence alignment and taxonomic classification in both QIIME and mothur [7] [86]. |
| GreenGenes (GG) Database | A 16S rRNA gene reference database that provides a taxonomy and multiple sequence alignment. | A historical standard for classification; compared against SILVA for performance [34] [7]. |
| Mock Community | A synthetic sample composed of genomic DNA from known microbial strains at defined ratios. | Serves as a critical control to benchmark pipeline performance, evaluate error rates, and optimize parameters like clustering threshold [34] [16]. |
| Oligos File | A text file containing barcode, primer, and linker sequences for demultiplexing and trimming. | Used in the make.contigs() command in mothur to correctly assign sequences to samples and remove primer sequences [46]. |
Addressing Primer Bias and Selection for Improved Taxonomic Resolution
In 16S rRNA gene sequencing, primer bias is a systematic error introduced during PCR amplification when primer sequences preferentially bind to and amplify specific taxonomic groups over others. This bias stems from sequence mismatches between the universal primer and the target DNA, leading to a distorted view of the microbial community structure and diversity [37]. The taxonomic resolution—the ability to distinguish between closely related microbial taxa—is highly dependent on the selected primer pair and the variable region(s) of the 16S rRNA gene it targets [34] [88].
The intergenomic variation present even within conserved regions of the 16S rRNA gene challenges the assumption of true "universal" primers [37]. This variation can lead to significant under-detection or complete omission of specific bacterial taxa, ultimately compromising the biological interpretation of microbiome data. Addressing primer bias is therefore not merely a technical refinement but a fundamental requirement for generating accurate, reproducible, and biologically meaningful results in microbial ecology, including within the QIIME and mothur analysis frameworks [7] [28].
Primer Performance: A Quantitative Comparison
Systematic evaluations of primer sets reveal substantial differences in their coverage and specificity, which directly impact taxonomic classification outcomes.
Table 1: In-silico Coverage of Primer Sets Across Dominant Gut Phyla
| Primer Set Identifier | Target Region | Coverage Threshold (≥70% across 4 phyla) | High Genus-Level Coverage (≥90% for ≥4 genera) | Key Findings |
|---|---|---|---|---|
| V3_P3 [37] | V3 | Achieved | Achieved | One of three balanced performers for core gut microbiome |
| V3_P7 [37] | V3 | Achieved | Achieved | Balanced coverage and specificity across key genera |
| V4_P10 [37] | V4 | Achieved | Achieved | Demonstrated robust phylum and genus-level coverage |
Table 2: Comparative Taxonomic Outcomes from Different Primer Pairs and Analysis Tools
| Factor | Impact on Taxonomic Resolution | Evidence |
|---|---|---|
| Primer Pair Choice | Different V-regions yield primer-specific clustering, affecting genus-level resolution more than phylum-level [34]. Specific taxa (e.g., Verrucomicrobia) may be missed by certain primers [34]. | Analysis of human stool samples showed donor samples clustered by primer pair rather than by donor when using different V-regions [34]. |
| Software & Database | mothur typically clusters more OTUs than QIIME, especially with GreenGenes database, affecting richness estimates [7]. SILVA database produces more comparable results between tools [7]. | In rumen microbiota, mothur assigned OTUs to a larger number of genera in low abundance (<10% RA) compared to QIIME when using GreenGenes [7]. |
| Multi-Primer Strategy | Mitigates bias by capturing a more comprehensive diversity; no single "universal" primer exists [37]. | In-silico analysis of 57 primer sets revealed significant limitations in widely used "universal" primers [37]. |
Experimental Protocols for Primer Selection and Validation
Protocol 1: In-silico Primer Evaluation and Selection
Purpose: To computationally evaluate and select optimal 16S rRNA primer sets for a specific microbiome study, such as the human gut, using a curated reference database.
Materials:
- Reference Database: SILVA SSU Ref NR database (release 138.1 or newer) [37].
- Software Tools: TestPrime 1.0 [37] or mopo16S [38].
- Primer Set List: A compiled list of candidate primer sequences from literature [37].
Method:
- Compile Candidate Primers: Systematically review literature and resources like the probeBase 16S primers database to compile a comprehensive list of candidate primer pairs targeting various variable regions (V1-V9) [37] [38].
- Retrieve and Curbate Reference Sequences: Download the SILVA SSU Ref NR 16S rRNA gene database. Filter sequences by length (>1,200 bp for Bacteria/Eukaryota, >900 bp for Archaea) to ensure quality.
- Perform In-silico PCR: Use TestPrime 1.0 to analyze each primer pair against the database. The analysis should allow for degeneracy in the primer sequences but permit no mismatches outside of these degenerate positions [37].
- Calculate Coverage: For each primer pair, calculate coverage as the percentage of eligible sequences from the target phyla (e.g., Actinobacteriota, Bacteroidota, Firmicutes, Proteobacteria for gut studies) that are successfully amplified in-silico [37].
- Apply Selection Criteria:
- Select primer pairs achieving ≥70% coverage across all four target phyla.
- From these, identify final candidate primers that also achieve ≥90% coverage for at least four out of ~20 representative genera of the core microbiome under investigation [37].
Protocol 2: Experimental Validation with Mock Communities
Purpose: To empirically validate the performance of candidate primer sets using a mock microbial community with a known composition.
Materials:
- Mock Community: ZymoBIOMICS Gut Microbiome Standard (D6331, Zymo Research). This community contains 19 bacterial and archaeal strains, providing a complex and defined benchmark [37].
- Laboratory Reagents: Standard reagents for DNA extraction and PCR amplification.
- Sequencing Platform: Illumina MiSeq or similar platform for amplicon sequencing.
Method:
- DNA Extraction: Extract genomic DNA from the mock community standard according to the manufacturer's instructions.
- PCR Amplification: Amplify the 16S rRNA gene from the extracted DNA using each of the candidate primer sets selected from Protocol 1. Perform PCR in triplicate to account for technical variability.
- Library Preparation and Sequencing: Prepare amplicon libraries following standard protocols and sequence on an Illumina MiSeq platform.
- Bioinformatic Analysis: Process the raw sequencing data using a standardized pipeline (e.g., QIIME 2 or mothur) to obtain taxonomic classifications [7] [28].
- Performance Assessment: Compare the observed taxonomic profile from each primer set to the known composition of the mock community. Calculate metrics such as:
Integrated Workflow for Mitigating Primer Bias
The following workflow integrates in-silico and experimental validation to inform primer selection and data analysis within a bioinformatics pipeline.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Primer Evaluation and Taxonomic Classification
| Resource Name | Type | Function in Addressing Primer Bias & Classification |
|---|---|---|
| SILVA SSU Ref NR [7] [37] | Reference Database | Curated, high-quality alignment of 16S/18S rRNAs; recommended for taxonomy assignment to reduce discrepancies between analysis tools. |
| ZymoBIOMICS Gut Microbiome Standard [37] | Mock Community | Defined mix of 19 bacterial & archaeal strains; gold standard for empirical validation of primer performance and bioinformatics pipeline accuracy. |
| TestPrime 1.0 [37] | Software Tool | Performs in-silico PCR against reference databases (e.g., SILVA) to predict primer coverage and identify biases before wet-lab experiments. |
| mopo16S [38] | Software Tool | Employs multi-objective optimization to design primer-set-pairs that maximize coverage, efficiency, and minimize amplification bias. |
| UNITE Database [89] | Reference Database | Curated database for fungal ITS sequences; used for analogous primer bias and taxonomy assignment challenges in fungal community studies. |
| CONSTAX [89] | Software Tool | Generates a consensus taxonomy from multiple classifiers (RDP, UTAX, SINTAX), improving the power and accuracy of taxonomic assignments. |
Primer bias is an inherent and significant challenge in 16S rRNA gene sequencing that cannot be ignored. A systematic, multi-faceted approach is required for robust taxonomic resolution. Key to this is the move away from relying on a single "universal" primer set and towards a strategy that employs in-silico screening and experimental validation with mock communities to select the most appropriate primers for a given study system [37]. Furthermore, the consistent use of curated reference databases like SILVA and the application of consensus classification tools can enhance the accuracy and reproducibility of taxonomic assignments downstream [7] [89]. By integrating these practices into standard QIIME and mothur pipelines, researchers can significantly mitigate the distortions caused by primer bias, leading to more reliable and insightful characterizations of microbial ecosystems.
Within 16S rRNA gene sequencing analysis, the choice of reference database is a critical methodological decision that directly impacts taxonomic assignment accuracy, diversity estimates, and ultimately, biological interpretation. Despite the availability of multiple curated databases, researchers often default to familiar options without systematic evaluation of their suitability for specific microbiome habitats. This application note examines the technical and performance distinctions between two widely used databases—SILVA and Greengenes—and provides evidence-based guidance for their implementation in microbial ecology studies, particularly for complex, non-human microbiomes.
Comparative evaluations consistently demonstrate that database selection introduces significant variation in taxonomic profiles, especially at finer taxonomic resolutions. This protocol synthesizes findings from multiple controlled assessments to establish why SILVA frequently outperforms Greengenes in specific microbial habitats and provides a framework for database selection within standard bioinformatics pipelines.
Comparative Database Performance Analysis
Key Characteristics and Comparative Metrics
Table 1: Fundamental characteristics and comparative performance of SILVA versus Greengenes.
| Feature | SILVA | Greengenes |
|---|---|---|
| Latest Update Status | Regularly updated [72] [90] | No updates since 2013 [70] [91] |
| Taxonomic Coverage | Bacteria, Archaea, Eukaryota [72] [90] | Bacteria and Archaea [70] |
| Species-Level Annotations | Contains some strain information without species designation [70] | Poor species-level annotation, many missing [70] |
| Genus-Level Accuracy (Mock Community) | ~35 genera identified, but highest false-positive rate (~20%) [70] | ~30 genera identified (lowest recovery) [70] |
| Impact on Alpha Diversity | Overestimates richness, underestimates evenness [70] | Overestimates richness, underestimates evenness [70] |
| Rumen Microbiota Performance | Preferred, produces comparable results between QIIME and mothur [7] [8] | Higher discrepancy between tools, lower sensitivity [7] [8] |
| Chicken Cecal Microbiota | Better resolution of Lachnospiraceae into genera [91] | Groups diverse Lachnospiraceae as "unclassified" [91] |
Experimental Validation Data
Table 2: Summary of database performance assessment using a mock community (59 uniformly distributed strains) [70].
| Performance Metric | SILVA | Greengenes | EzBioCloud |
|---|---|---|---|
| True Positive Genera (avg) | ~35 | ~30 | >40 |
| False Positive Genera (avg) | ~20% | Moderate | Lowest |
| Genus-Level Resolution | Sufficient | Poor | Best |
| Species-Level Resolution | ~25 correct species | Few correct species | ~40 correct species |
Independent validation in specialized microbiomes reinforces these trends. In chicken cecal studies, SILVA provided superior resolution of the family Lachnospiraceae into separate genera, whereas Greengenes grouped these members into a single "unclassified" category [91]. This enhanced resolution directly translated to more biologically informative linear discriminant analysis, with SILVA identifying more differentially abundant genera [91].
For rumen microbiota analysis, which contains numerous uncultured species, SILVA produced more consistent results between QIIME and mothur platforms, whereas Greengenes introduced significant tool-specific variation, particularly for low-abundance microorganisms [7] [8].
Wet-Lab and Computational Protocols
Experimental Workflow for Database Comparison
The following workflow provides a standardized approach for evaluating database performance with experimental or mock community data:
Detailed Protocol Steps
Wet-Lab Procedures (Wet-Bench Protocol)
Mock Community Preparation:
- Obtain defined mock community with known composition (e.g., ZymoBIOMICS or in-house standardized community)
- Extract DNA using standardized extraction kit (e.g., DNeasy PowerSoil Pro Kit)
- Amplify target hypervariable region (V3-V4 recommended) using dual-indexing strategy
- Perform library preparation with Illumina-compatible protocols
- Sequence on appropriate Illumina platform (MiSeq or NovaSeq) to achieve minimum 50,000 reads per sample
Sample Processing:
- Process experimental samples alongside mock community controls using identical laboratory protocols
- Include extraction blanks and PCR negatives to monitor contamination
- Pool libraries in equimolar ratios based on fluorometric quantification
Bioinformatics Implementation (Dry-Lab Protocol)
Quality Control Processing:
- For QIIME2: Use
q2-dada2for denoising, quality filtering, and chimera removal with parameters:--p-trunc-len 0 --p-trim-left 0 --p-max-ee 2.0 - For mothur: Implement
make.contigs(),screen.seqs(),filter.seqs(),chimera.vsearch(), andcluster.split()following standard SOP - Remove sequences with ambiguous bases, homopolymers >8bp, and quality scores
Taxonomic Assignment:
- Download latest SILVA release (SSU Ref NR 99) and Greengenes (13_8) reference files formatted for your pipeline
- For QIIME2: Use
feature-classifier classify-sklearnwith pre-trained classifiers - For mothur: Use
classify.seqs()method with Wang algorithm and 80% bootstrap confidence threshold - Remove chloroplast, mitochondrial, and eukaryotic sequences from subsequent analysis
Comparative Analysis:
- Generate taxonomic composition tables for both database assignments
- Calculate alpha diversity indices (Chao1, Shannon, Simpson) and beta diversity metrics (Bray-Curtis, UniFrac)
- For mock community: Calculate precision, recall, and F1-score against known composition
- For biological samples: Perform differential abundance analysis (LEfSe, ANCOM-BC) to identify database-specific biases
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key reagents, databases, and computational tools for 16S rRNA database evaluation.
| Resource | Specification/Version | Primary Function |
|---|---|---|
| SILVA Database | Release 138.1 or newer [90] | Comprehensive taxonomic reference for Bacteria, Archaea, Eukarya |
| Greengenes Database | Version 13_8 [70] [92] | Bacterial and Archaeal reference (historically used) |
| QIIME2 Platform | 2024.5 or newer [90] [7] | Integrated microbiome analysis pipeline |
| mothur Platform | 1.48.0 or newer [7] [93] | 16S rRNA sequence processing and analysis |
| Mock Community | ZymoBIOMICS or in-house [70] | Validation of database classification accuracy |
| DADA2 Plugin | QIIME2 implementation [90] | Amplicon Sequence Variant (ASV) inference |
| Naive Bayes Classifier | q2-feature-classifier [90] [94] | Taxonomic assignment algorithm |
| RDP Training Set | Version 18 [92] | Alternative reference dataset for comparison |
Discussion and Implementation Guidelines
Database Selection Framework
The collective evidence indicates that SILVA is generally preferred for microbiome studies requiring genus-level resolution, particularly for complex environments like rumen and gut ecosystems [7] [8] [91]. SILVA's comprehensive taxonomy, regular updates, and superior resolution of challenging groups like Lachnospiraceae make it particularly valuable for hypothesis-driven research requiring taxonomic precision [91].
Greengenes remains suitable for method comparison studies or reproducing earlier analyses, but its outdated taxonomy (2013) introduces increasing limitations [70] [91]. The lack of updates means novel taxa discovered in the past decade are absent, potentially misclassifying emerging microorganisms of interest.
Emerging Alternatives and Future Directions
While this protocol focuses on SILVA versus Greengenes, researchers should consider emerging unified resources. The Greengenes2 database represents a significant advancement by integrating genomic and 16S rRNA data within a consistent phylogenetic framework [94]. Additionally, manually curated integrated databases like GSR-DB (Greengenes, SILVA, RDP) show promising results for species-level resolution by addressing nomenclature inconsistencies [92].
Concluding Recommendations
For robust microbiome analysis, researchers should:
- Validate database choice using mock communities when possible
- Prioritize SILVA for studies requiring maximum taxonomic resolution
- Document database versions thoroughly to ensure reproducibility
- Consider database integration approaches when analyzing novel or undersampled environments
- Align database selection with specific research questions rather than default preferences
This systematic approach to database selection ensures that taxonomic assignments support accurate biological interpretation rather than introducing technical artifacts that might compromise study conclusions.
The analysis of low microbial biomass environments—such as certain human tissues (blood, placenta, respiratory tract), treated drinking water, hyper-arid soils, and the deep subsurface—poses unique challenges for 16S rRNA gene sequencing studies [95]. In these environments, the microbial DNA signal approaches the limits of detection of standard DNA-based sequencing approaches, making results disproportionately vulnerable to contamination from external sources [95] [23]. The inevitability of contamination becomes a critical concern when working near detection limits, as even minute amounts of contaminant DNA can strongly influence study results and their interpretation, potentially leading to false conclusions about microbial presence, ecological patterns, or evolutionary signatures [95].
The fundamental issue lies in the proportional nature of sequence-based datasets. In high-biomass samples like human stool or surface soil, the target DNA "signal" substantially exceeds the contaminant "noise." In contrast, low-biomass samples may contain contaminant DNA at levels comparable to or even exceeding the true biological signal, creating misleading representations of microbial community composition [95] [96]. This problem is particularly acute in 16S rRNA amplicon sequencing studies, where contaminants can originate from multiple sources including human operators, sampling equipment, laboratory reagents, kits, and cross-contamination between samples during processing [95]. Research indicates that despite widespread awareness of these issues, the use of appropriate controls has not increased over the past decade, maintaining justifiable skepticism toward published microbiome studies in low-biomass systems [95].
In low-biomass 16S rRNA studies, contamination can be introduced at virtually every stage of the experimental workflow, from sample collection through data analysis [95]. The major sources of contamination include:
- Human operators: Microbial cells and DNA shed from skin, hair, clothing, or generated through breathing and talking can contaminate samples [95]. One study noted that human aerosol droplets generated while breathing or talking represent a significant contamination risk [95].
- Sampling equipment: Collection vessels, swabs, and tools may harbor microbial DNA even after sterilization [95]. It is crucial to distinguish that sterility (absence of viable cells) is not equivalent to being DNA-free, as cell-free DNA can persist on surfaces even after autoclaving or ethanol treatment [95].
- Laboratory reagents and kits: Commercial DNA extraction kits, PCR reagents, and water often contain trace amounts of bacterial DNA that can be amplified and sequenced [95] [23]. These kit-borne contaminants can become the dominant signal in low-biomass samples.
- Cross-contamination between samples: Well-to-well leakage during PCR setup or other liquid handling procedures can transfer DNA between samples, a phenomenon particularly problematic when high-biomass and low-biomass samples are processed together [95] [96]. This well-to-well contamination is a common form of cross-contamination where biological samples leak into controls [96].
- Laboratory environments: Airborne particles and surfaces in laboratory facilities can serve as reservoirs of contaminating DNA [95].
Impact on Data Quality and Interpretation
The consequences of contamination in low-biomass 16S rRNA studies are severe and multifaceted. Even small amounts of contaminant DNA can distort ecological patterns and diversity metrics, leading to incorrect biological interpretations [95]. In clinical diagnostics, contamination can cause false attribution of pathogen exposure pathways or misdiagnosis of infections [95]. The ongoing debate surrounding the 'placental microbiome' exemplifies how contamination issues can fuel scientific controversy, with some studies potentially misattracting contaminant DNA as authentic signal [95].
Different types of contamination present distinct challenges. Environmental contamination from reagents or laboratory environments introduces consistent contaminant taxa across samples, while cross-contamination between samples creates variable contamination patterns that can be particularly difficult to distinguish from true biological signal [96]. The problem is compounded by the fact that practices suitable for handling higher-biomass samples may produce misleading results when applied to low microbial biomass samples [95].
Best Practices for Contamination Prevention
Pre-Analytical Phase: Sample Collection and Handling
Table 1: Strategies for Preventing Contamination During Sample Collection and Processing
| Stage | Practice | Implementation |
|---|---|---|
| Sample Collection | Decontaminate sources of contaminant cells or DNA | Use 80% ethanol followed by nucleic acid degrading solution (e.g., bleach, UV-C light) [95] |
| Use personal protective equipment (PPE) | Wear gloves, goggles, coveralls/cleansuits, and shoe covers to limit human-derived contamination [95] | |
| Use single-use DNA-free collection materials | Employ pre-sterilized swabs and collection vessels that remain sealed until sample collection [95] | |
| Sample Storage | Immediate preservation | Freeze samples at -20°C or -80°C as quickly as possible; use preservation buffers if immediate freezing isn't possible [1] |
| Aliquot samples | Avoid repeated freeze-thaw cycles by creating single-use aliquots [1] | |
| Laboratory Processing | Dedicated workspace | Use separate areas for pre- and post-PCR activities, preferably with UV laminar flow hoods [95] |
| Reagent verification | Check that preservation solutions and reagents are DNA-free; use ultra-pure reagents specifically certified for microbiome work [95] |
Implementing rigorous contamination control during sample collection is paramount. Before sampling, researchers should conduct test runs to identify and reduce potential contamination sources [95]. During sampling, personnel should receive comprehensive training to ensure procedures are followed consistently, with particular attention to minimizing sample handling and exposure to potential contamination sources [95]. For equipment decontamination, sodium hypochlorite (bleach), UV-C exposure, hydrogen peroxide, ethylene oxide gas, or commercially available DNA removal solutions are recommended where safe and practical, as these effectively remove DNA rather than just viable cells [95].
Experimental Design and Controls
The inclusion of appropriate controls is fundamental for identifying contamination sources and interpreting results in context. Different control types serve distinct purposes in low-biomass studies:
- Negative controls (also called extraction controls or no-template controls): These include empty collection vessels, aliquots of preservation solution, or swabs exposed to the sampling environment air [95] [23]. They should be processed alongside actual samples through all stages, including DNA extraction and sequencing, to identify contaminants introduced during laboratory processing [95].
- Positive controls: Mock communities with known compositions of bacterial strains help assess sequencing quality, PCR amplification efficiency, and bioinformatics performance [23] [97].
- Sampling controls: Swabs of PPE, sampling surfaces, or sampling fluids help identify specific contamination sources [95]. For example, in a fetal meconium study, researchers swabbed decontaminated maternal skin before the procedure and used additional swabs exposed to operating theatre air to identify contamination sources [95].
Multiple controls should be included throughout the experiment to accurately quantify the nature and extent of contamination, enabling informed decisions during data analysis about which sequences likely represent contaminants [95]. The distribution of samples and controls on sequencing plates should also be randomized to avoid systematic bias from plate position effects.
Wet Lab Protocols for Low-Biomass Samples
DNA Extraction and Library Preparation
When processing low-biomass samples in the laboratory, specific modifications to standard protocols are necessary:
DNA Extraction Optimization:
- Utilize mechanical lysis methods (e.g., bead beating) appropriate for the sample type to ensure efficient cell disruption [1].
- Include negative extraction controls (reagents only) with every batch of samples to monitor kit-borne contamination [23].
- Consider using extraction kits specifically designed for low-biomass samples or implementing additional purification steps to remove inhibitors.
- Evaluate different input volumes and elution volumes to maximize DNA yield while minimizing co-extraction of inhibitors.
16S rRNA Gene Amplification:
- Optimize PCR cycle numbers to minimize amplification bias while ensuring sufficient product for library preparation [97]. Excessive cycling can amplify contaminating DNA and increase stochastic effects.
- Use high-fidelity DNA polymerases to reduce amplification errors.
- Employ minimal template volumes in PCR reactions to reduce introduction of contaminants.
- Include multiple negative PCR controls (water instead of template) to identify contamination introduced during amplification.
Library Preparation:
- Implement rigorous cleaning steps using magnetic beads or other purification methods to remove primers, primer dimers, and other contaminants [1].
- Use unique barcodes for each sample to enable multiplexing while maintaining sample identity [1].
- Quantify libraries using sensitive methods such as fluorometry rather than spectrophotometry to accurately assess DNA concentration while detecting potential contamination.
Incorporating Internal Controls for Quantification
For absolute quantification in low-biomass samples, incorporating internal controls provides significant advantages:
Spike-In Controls:
- Add known quantities of synthetic or foreign DNA sequences not found in the samples of interest to serve as internal standards [97].
- Use spike-ins at different concentrations across samples to create standard curves for absolute abundance estimation [97].
- Select spike-in organisms that are phylogenetically distinct from expected communities but amplify efficiently with the same primers.
Mock Communities:
- Include commercially available mock community standards with known bacterial composition and abundance [97].
- Process mock communities alongside experimental samples to monitor technical variability and quantify biases in amplification and sequencing.
- Use mock community data to normalize samples and correct for technical artifacts.
The use of internal controls enables transformation of relative abundance data into absolute quantification, addressing a key limitation of amplicon sequencing for low-biomass applications where microbial load is biologically relevant [97].
Bioinformatics Decontamination Strategies
Decontamination Tools and Approaches
Table 2: Bioinformatics Tools for Decontaminating 16S rRNA Data from Low-Biomass Samples
| Tool | Method Category | Key Features | Applicability |
|---|---|---|---|
| micRoclean [96] | Control-based, Sample-based | Two pipelines: Original Composition Estimation (leverages SCRuB) and Biomarker Identification; provides filtering loss statistic | Flexible tool for low-biomass studies with guidance on pipeline selection based on research goals |
| decontam [96] | Control-based, Sample-based | Identifies contaminant features based on prevalence in negative controls or association with DNA concentration | Widely used; integrates with QIIME and R pipelines |
| SCRuB [96] | Control-based | Models and removes contamination, including well-to-well leakage; can account for spatial relationships on sequencing plates | Ideal when well location information is available |
| MicrobIEM [96] | Control-based | Removes only proportion of features identified as contamination rather than entire features | Useful for partial decontamination |
| microDecon [96] | Control-based | Uses negative controls to subtract contaminant reads from samples | Straightforward subtraction-based approach |
Bioinformatics decontamination methods broadly fall into three categories: (1) Blocklist methods that remove features previously identified in the literature as common contaminants; (2) Sample-based methods that identify contaminant features based on their abundance patterns across samples or batches; and (3) Control-based methods that leverage negative controls to identify contaminant sequences [96]. The most effective approaches often combine multiple strategies.
The micRoclean package, specifically designed for low-biomass studies, offers two distinct pipelines with guidance on selection based on research goals [96]. The "Original Composition Estimation" pipeline aims to closely estimate the original microbiome composition prior to contamination and is ideal when concerned about well-to-well contamination with available well location information [96]. The "Biomarker Identification" pipeline strictly removes all likely contaminant features to minimize the impact of contaminants on downstream biomarker analyses [96].
Implementation in QIIME 2 and mothur Pipelines
QIIME 2 Implementation:
- After generating the feature table and representative sequences, integrate decontamination tools such as decontam or microDecon.
- Use the q2-quality-control plugin to compare samples against negative controls.
- Filter features identified as contaminants before downstream diversity analyses and taxonomy assignment.
- Apply prevalence-based or frequency-based methods depending on the availability of sample metadata.
mothur Implementation:
- Incorporate decontamination steps after the contig assembly and before the classification steps.
- Use the remove.lineage command to exclude taxa previously identified as common contaminants.
- Implement cross-sample comparison methods to identify outliers potentially representing contaminants.
- Leverage the split.groups function to analyze controls separately from samples.
Both pipelines benefit from the calculation of a filtering loss (FL) statistic to quantify the impact of contaminant removal on the overall covariance structure of the data [96]. This metric, calculated as FLJ = 1 - (||YTY||F2/||XTX||F2), where X is the pre-filtering count matrix and Y is the post-filtering count matrix, helps prevent over-filtering that might remove legitimate biological signal [96]. Values closer to 0 indicate low contribution of removed features to overall covariance, while values closer to 1 could indicate over-filtering [96].
Experimental Workflow and Data Analysis
Comprehensive Workflow Diagram
The following diagram illustrates the integrated experimental and computational workflow for low-biomass 16S rRNA studies, incorporating contamination controls at each stage:
Low-Biomass 16S rRNA Study Workflow
Data Interpretation Guidelines
Interpreting results from low-biomass 16S rRNA studies requires careful consideration of contamination risks:
- Compare with controls: Authentic signals should be substantially more abundant in samples than in negative controls [95]. Features present at similar levels in both samples and negative controls likely represent contamination.
- Assess taxonomic plausibility: Consider whether identified taxa are biologically plausible for the sample type. Unexpected environmental bacteria or common reagent contaminants should be treated with skepticism.
- Evaluate abundance patterns: True biological signals often show structured variation across sample groups, while contaminants may appear randomly distributed.
- Apply multiple lines of evidence: Combine evidence from negative controls, prevalence across samples, taxonomic identity, and prior biological knowledge when assessing signal authenticity.
- Report contamination transparency: Clearly document all controls used, contamination removal methods applied, and the proportion of sequences removed during decontamination steps [95].
Essential Research Reagent Solutions
Table 3: Key Research Reagents and Materials for Low-Biomass 16S rRNA Studies
| Reagent/Material | Function | Application Notes |
|---|---|---|
| DNA-free collection swabs | Sample collection without introducing contaminant DNA | Pre-sterilized and certified DNA-free; single-use only [95] |
| Nucleic acid removal solutions | Decontaminate surfaces and equipment | Sodium hypochlorite (bleach), UV-C light, hydrogen peroxide, or commercial DNA removal solutions [95] |
| DNA extraction kits for low biomass | Maximize DNA yield while minimizing contamination | Select kits with minimal microbial DNA background; include bead-beating for mechanical lysis [97] [1] |
| Mock community standards | Positive controls for sequencing and quantification | Commercially available standards with known composition (e.g., ZymoBIOMICS standards) [97] |
| Spike-in controls | Internal standards for absolute quantification | Foreign DNA not found in samples; added at known concentrations [97] |
| Ultra-pure molecular biology reagents | PCR and library preparation with minimal contaminant DNA | Specifically certified for microbiome studies; lot-testing recommended [95] |
| DNA-free plasticware and tubes | Sample processing without introducing contaminants | Certified DNA-free; sterilized by autoclaving or UV treatment [95] |
The study of low-biomass environments using 16S rRNA sequencing presents distinctive challenges that demand rigorous contamination control throughout the entire research workflow, from experimental design through data interpretation. Success in this field requires integrated approach combining careful laboratory practices, appropriate controls, and sophisticated bioinformatics tools specifically validated for low-biomass applications. By implementing the comprehensive strategies outlined in this protocol—including proper sample handling, systematic use of controls, computational decontamination, and transparent reporting—researchers can significantly improve the reliability and interpretability of their low-biomass microbiome studies. As the field continues to evolve, adoption of these best practices will be essential for generating robust, reproducible results that advance our understanding of microbial communities in low-biomass environments.
Operating System Compatibility and Performance
The choice of operating system (OS) can influence the execution and results of 16S rRNA analysis pipelines, though the extent varies between tools.
Table 1: Operating System Support and Dependencies for Bioinformatics Pipelines
| Pipeline | OS Compatibility | Core Language | Installation Complexity | Key Dependencies | OS-Induced Result Variation |
|---|---|---|---|---|---|
| QIIME 2 | Linux, macOS | Python | High (dependency management) | Multiple external tools & libraries | Minimal (Outputs identical on Linux and macOS) [86] |
| mothur | Linux, macOS, Windows | C/C++ | Low (Standalone executable) | Self-contained; minimal external dependencies | Minimal (Outputs nearly identical on Linux and macOS) [86] |
| Kraken 2/Bracken | Linux, macOS | C++ | Moderate | Requires database building | Information not specified in search results |
| Bioconductor | Linux, macOS, Windows | R | Moderate | R package ecosystem | Minimal (Outputs identical on Linux and macOS) [86] |
| UPARSE | Linux, macOS | C++ | Moderate | Information not specified in search results | Minimal (Outputs nearly identical on Linux and macOS) [86] |
Evidence indicates that for major pipelines like QIIME2, Bioconductor, UPARSE, and mothur, the choice between Linux and Mac OS introduces only minimal to non-existent differences in taxonomic classification results, enhancing the reproducibility and comparability of studies conducted on different standard operating systems [86].
Computational Resource Requirements and Performance
The underlying architecture and algorithms of bioinformatics tools directly impact their computational efficiency, including processing speed and memory usage.
Table 2: Computational Resource and Performance Benchmarking
| Pipeline | Computational Architecture | Speed | Memory (RAM) Usage | Key Strengths | Noted Limitations |
|---|---|---|---|---|---|
| QIIME 2 | Wrapper for multiple tools | Slower (Most computationally expensive) [98] | High (~100x more RAM than Kraken 2) [98] | High accuracy, extensive plugin ecosystem | High computational cost can be prohibitive for large datasets [98] |
| mothur | Self-contained, compiled C/C++ | Fast (e.g., align.seqs 21.9x faster than PyNAST) [6] | Moderate | Standalone nature, OS-independent code, high speed [6] | Fewer code contributions from community due to C++ [6] |
| Kraken 2/Bracken | Alignment-free, k-mer based | Ultrafast (Up to 300x faster than QIIME 2) [98] | Low (~100x less RAM than QIIME 2) [98] | Exceptional speed and memory efficiency, accurate per-read assignments | Requires a specialized database building step [98] |
| ASV Algorithms (DADA2, Deblur) | Denoising-based | Varies | Varies | Single-nucleotide resolution, reproducible ASVs across studies | Can suffer from over-splitting of 16S rRNA gene copies [99] |
| OTU Algorithms (UPARSE, mothur) | Clustering-based (e.g., 97% identity) | Varies | Varies | Lower error rates, robust to sequencing noise | Can suffer from over-merging of distinct biological sequences [99] |
Experimental Protocols for Benchmarking Pipelines
To ensure reproducible and robust microbiome analysis, following standardized protocols for benchmarking and validation is crucial.
Protocol for Cross-Platform Validation
Objective: To verify that a bioinformatics pipeline produces consistent results across different operating systems.
- Sample Preparation: Use a well-characterized dataset, such as a mock community with known composition or a representative subset of real 16S rRNA sequencing data.
- Software Installation: Install the same version of the target pipeline (e.g., QIIME2, mothur) on a Linux and a macOS machine. For QIIME2, use the recommended installation method (e.g., conda environment). For mothur, download the pre-compiled executable for each OS.
- Analysis Execution: Run the exact same analysis workflow, including all parameters and reference databases (e.g., SILVA 132), on both systems.
- Output Comparison:
- Compare the final taxonomic abundance tables (e.g., at the genus level) using statistical tests such as the Friedman rank sum test [86].
- Assess diversity metrics (e.g., alpha and beta diversity indices) generated from both outputs.
- Expected Outcome: For pipelines like QIIME2 and Bioconductor, taxonomic assignments and relative abundances should be identical. For others like UPARSE and mothur, only minimal, non-significant differences are acceptable [86].
Protocol for Performance Benchmarking
Objective: To evaluate the computational efficiency and resource consumption of different pipelines.
- Test Environment: Establish a controlled computing environment with dedicated resources (e.g., a single server or virtual machine) to ensure consistent measurement.
- Dataset Selection: Select a standardized 16S rRNA dataset (e.g., from the Mockrobiota database [99] or simulated reads from known communities [98]).
- Metric Measurement:
- CPU Time: Measure the total wall time and CPU time required for the pipeline to complete from raw reads to taxonomic table.
- Peak Memory Usage: Monitor the maximum RAM used during the pipeline's execution.
- Storage: Note the disk space required for intermediate files and final outputs.
- Execution: Run each pipeline (e.g., QIIME 2's
q2-feature-classifier, Kraken 2, mothur) on the same dataset, ensuring the same level of taxonomic resolution and output format for a fair comparison. Repeat the runs to account for variability. - Data Analysis: Compile the metrics into a comparative table (see Table 2). The significant speed (up to 300x faster) and memory (~100x less RAM) advantages of tools like Kraken 2 over QIIME 2 will be evident from this analysis [98].
Workflow Diagram of Computational Considerations
The following diagram synthesizes the key computational factors and their interrelationships in selecting and deploying a 16S rRNA analysis pipeline.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Computational Materials for 16S rRNA Pipeline Analysis
| Item Name | Function / Purpose | Example Sources / Specifications |
|---|---|---|
| Mock Microbial Community | A DNA sample composed of genomic material from known bacterial strains. Serves as a ground truth for validating pipeline accuracy and benchmarking performance. | HC227 (227 strains), Mockrobiota database samples [99] |
| Reference Taxonomy Database | Curated collections of 16S rRNA sequences with taxonomic labels. Essential for assigning taxonomy to unknown sequence reads. | SILVA, Greengenes, RDP [7] [98] |
| High-Performance Computing (HPC) Infrastructure | Provides the necessary computational power (CPU, large RAM, fast storage) to run resource-intensive pipelines in a reasonable time. | Local servers, cloud computing instances (AWS, GCP), institutional HPC clusters |
| Containerization Platforms | Technology that packages a pipeline and its dependencies into a single, portable unit, ensuring reproducibility across different computing environments. | Docker, Singularity (particularly for HPC) |
| Pre-compiled Bioinformatics Binaries | Ready-to-run executable versions of software, avoiding the need for compilation and simplifying installation, especially for tools written in C/C++. | mothur executables, USEARCH [6] |
| Standardized Data Formats | Agreed-upon file formats for storing sequencing data and results, enabling interoperability between different pipelines and tools. | FASTQ (raw reads), BIOM (taxonomic table), FASTA (sequences) |
The accurate profiling of low-abundance microbial taxa represents a significant challenge in 16S rRNA amplicon sequencing studies. While conventional bioinformatics pipelines provide robust tools for community analysis, their performance varies considerably when detecting and quantifying rare community members with relative abundance below 1-10% [7]. These low-abundance organisms, though numerically minor, can possess disproportionate ecological and clinical significance, functioning as keystone species, pathogens, or biomarkers for specific host conditions. The limitations of standard analytical approaches necessitate specialized strategies spanning experimental design, computational tool selection, and database curation to achieve reliable detection of these elusive taxa.
The fundamental challenges in low-biomass taxon detection stem from multiple sources, including sequencing artifacts, PCR amplification biases, database incompleteness, and algorithmic limitations in bioinformatics pipelines. Each stage of the analytical process—from primer selection to taxonomic classification—introduces potential biases that can either obscure genuine low-abundance signals or generate false positives. Within the context of QIIME and mothur pipelines, researchers must understand how parameters and reference databases influence sensitivity thresholds, particularly for complex microbiomes like the rumen where many species remain uncultivated and poorly represented in standard databases [7]. This protocol details evidence-based strategies to enhance detection capabilities for comprehensive microbiome characterization.
Comparative Performance of Bioinformatics Pipelines
Pipeline and Database Selection Impact
The choice of bioinformatics software and reference database significantly influences detection sensitivity for low-abundance taxa. A direct comparison of QIIME (v1.9.1) and mothur (v1.39.5) using dairy cow rumen microbiota revealed critical differences in performance characteristics, especially for taxa with relative abundance below 10% [7]. When analyzing identical 16S rRNA (V4 region) amplicon datasets, mothur consistently clustered sequences into a larger number of OTUs regardless of the reference database used, suggesting higher analytical sensitivity for rare organisms [7]. This difference in OTU clustering behavior directly impacted downstream diversity metrics and ecological interpretations.
The reference database selection proves equally crucial for detection sensitivity. The same study evaluated both GreenGenes (May 2013 version) and SILVA (release 132) databases, finding that database choice substantially moderated the differences between QIIME and mothur [7]. While both pipelines identified similar high-abundance genera (Bifidobacterium, Butyrivibrio, Methanobrevibacter, Prevotella, and Succiniclasticum) at relative abundance >1% regardless of database, significant differences emerged for less abundant community members [7]. Specifically, when using GreenGenes, mothur assigned OTUs to a larger number of genera and at higher relative abundances for low-frequency microorganisms, resulting in significantly richer observed communities (P < 0.05) and more favorable rarefaction curves [7]. These differences directly influenced beta diversity calculations, affecting how samples clustered in multivariate space and potentially leading to different biological conclusions.
Table 1: Comparison of QIIME and Mothur Performance with Different Reference Databases
| Metric | GreenGenes Database | SILVA Database |
|---|---|---|
| Number of OTUs clustered | Mothur > QIIME (P < 0.001) | Mothur > QIIME (P < 0.001) |
| Genera detected (RA > 0.1%) | Mothur: 29, QIIME: 24 | Differences attenuated |
| Unclassified OTUs at genus level | QIIME: 61%, Mothur: 67% | Similar patterns but reduced differences |
| Impact on beta diversity | Significant differences between tools | Differences reduced but not eliminated |
| Recommended for low-abundance taxa | SILVA preferred for both pipelines | SILVA preferred for both pipelines |
Enhanced Detection Through Multi-Region Sequencing
Conventional 16S rRNA amplicon sequencing typically targets 1-2 hypervariable regions, limiting phylogenetic resolution due to varying taxonomic discrimination power across different variable regions. Emerging approaches that leverage multiple variable regions significantly improve species-level classification, thereby enhancing detection confidence for low-abundance taxa [100]. The xGen 16S Amplicon Panel v2 enables amplification of all nine variable regions, while the complementary SNAPP-py3 bioinformatics pipeline facilitates analysis of this multi-region data [100].
This multi-region approach mitigates the resolution limitations inherent in single-region sequencing by providing substantially more phylogenetic information per read. Different variable regions exhibit varying discrimination power for specific taxonomic groups; by combining information across regions, classification ambiguity is reduced, especially for closely related species that may be present at low abundances [100]. Validation studies using mock communities demonstrate that this approach provides highly reproducible species-level identification, with technical replicates (both within-run and between-run) showing minimal variance in low-abundance taxon detection [100]. The protocol's effectiveness extends to challenging sample types like infant gut microbiomes, where low biomass and high interindividual variability complicate rare taxon detection.
Advanced Profiling Techniques
Strain-Level Resolution with Shotgun Metagenomics
For applications requiring strain-level tracking of low-abundance organisms, shotgun metagenomics with specialized computational tools offers superior resolution compared to 16S rRNA amplicon sequencing. ChronoStrain represents a significant advancement as a sequence quality- and time-aware Bayesian model specifically designed for profiling strains in longitudinal samples [101]. This approach explicitly models the presence or absence of each strain and produces probability distributions over abundance trajectories, making it particularly effective for low-biomass taxa that hover near detection thresholds.
ChronoStrain's performance advantages are especially pronounced in longitudinal study designs where temporal information provides additional constraints for strain detection. In benchmarking evaluations against alternative methods (StrainGST, StrainEst, mGEMS), ChronoStrain significantly outperformed competitors in both abundance estimation accuracy and presence/absence prediction for low-abundance strains [101]. The method's improved lower limit of detection was validated using paired sample isolates from the Baby Biome Study, where it demonstrated superior detection of Enterococcus faecalis strains in infant fecal samples [101]. Similarly, in studies of women with recurrent urinary tract infections, ChronoStrain provided improved interpretability for tracking Escherichia coli strain blooms in longitudinal fecal samples [101].
Table 2: ChronoStrain Performance Metrics for Low-Abundance Strain Detection
| Performance Metric | ChronoStrain | Timeseries-Agnostic Mode | Other Methods (StrainGST, StrainEst, mGEMS) |
|---|---|---|---|
| RMSE-log (low-abundance strains) | Significantly lower | Moderate | Higher |
| AUROC (presence/absence) | Superior | Good | Variable/Lower |
| Temporal resolution | High (longitudinal modeling) | None | Limited |
| Strain-level detection limit | Improved lower limit | Moderate | Higher |
| Runtime | Comparable | Comparable | Comparable |
Expanding Reference Databases with Metagenome-Assembled Genomes
Reference database incompleteness represents a fundamental limitation for detecting uncharacterized low-abundance taxa. MetaPhlAn 4 addresses this challenge by integrating metagenome-assembled genomes (MAGs) with microbial isolate genomes to create a substantially expanded reference framework [102]. This integration enables the definition of species-level genome bins (SGBs) for both known (kSGBs) and unknown (uSGBs) taxonomic groups, dramatically improving coverage of microbial diversity across environments.
The MetaPhlAn 4 approach clusters reference genomes and MAGs at 5% genomic distance to define SGBs, then identifies species-specific marker genes for profiling [102]. This strategy has expanded the database to include 26,970 SGBs with defined unique marker genes (21,978 kSGBs and 4,992 uSGBs) [102]. The practical impact is significant, with MetaPhlAn 4 explaining approximately 20% more reads in human gut microbiomes and over 40% more reads in less-characterized environments like the rumen microbiome compared to previous methods [102]. This enhanced reference database enables more comprehensive profiling of previously undetectable taxa, revealing uncharacterized species that serve as robust biomarkers for host conditions and lifestyles.
Experimental Protocols
Protocol 1: Optimized 16S rRNA Analysis for Low-Abundance Taxa
Sample Preparation and Sequencing
- Utilize the xGen 16S Amplicon Panel v2 for library preparation, which amplifies all nine variable regions of the 16S rRNA gene to maximize phylogenetic resolution [100].
- Include mock community controls (e.g., ZymoBIOMICS) containing known abundances of bacterial species to validate detection thresholds and quantify false positive/negative rates [100].
- Sequence on Illumina MiSeq platform with 250 bp paired-end chemistry to ensure sufficient read length for multiple variable region coverage.
Bioinformatics Processing
- Process raw sequences through the SNAPP-py3 pipeline, specifically designed for multi-region 16S data [100].
- For traditional single-region approaches, implement both QIIME (v1.9.1+) and mothur (v1.39.5+) in parallel to compare sensitivity thresholds [7].
- Apply strict quality filtering (Q-score >30) but avoid excessive read trimming to preserve phylogenetic information [103].
- Use the SILVA database (release 132+) for taxonomic classification, as it demonstrates superior performance for detecting low-abundance rumen microbiota compared to GreenGenes [7].
- Cluster sequences at 97% similarity for OTU picking, but retain rare OTUs (those with ≥2 reads) rather than applying abundance filters [7].
Sensitivity Enhancement Steps
- Normalize read counts using CSS (cumulative sum scaling) or TSS (total sum scaling) rather than rarefaction to preserve information from low-count samples [103].
- For differential abundance testing, apply specialized methods like DESeq2 or ANCOM that account for compositionality and handle sparse data effectively.
- Validate low-abundance taxa by checking for consistent detection across technical replicates and complementary variable regions.
Protocol 2: Strain-Resolved Tracking with ChronoStrain
Database Preparation
- Compile a custom database of marker sequence seeds, which can include MetaPhlAn core marker genes, sequence typing genes, fimbrial genes, and known virulence factors [101].
- Align seeds to reference genome databases, identifying sufficiently similar matches as marker sequences for corresponding genomes.
- Set strain clustering thresholds based on research questions, typically ranging from 99.8% to 100% sequence similarity for distinguishing distinct strains [101].
Longitudinal Sample Processing
- Process raw FASTQ files through ChronoStrain's initial bioinformatics steps, which filter reads against the custom marker database [101].
- Input filtered read files with quality scores, sample metadata with collection timepoints, and the custom marker database into the ChronoStrain Bayesian model.
- Run the model to obtain presence/absence probabilities and probabilistic abundance trajectories for each strain across timepoints.
Result Interpretation
- Focus on strains with presence probability >0.95 for high-confidence detections.
- Examine abundance trajectories for consistent patterns rather than single-timepoint abundances.
- Validate low-abundance strain detections against known clinical or phenotypic data where available.
Experimental Workflows
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Tool/Reagent | Function | Application Notes |
|---|---|---|
| xGen 16S Amplicon Panel v2 | Amplifies all 9 variable regions of 16S rRNA gene | Enhances species-level resolution compared to single-region approaches [100] |
| SNAPP-py3 Pipeline | Bioinformatics analysis of multi-region 16S data | Specifically designed for xGen panel output [100] |
| SILVA Database | Taxonomic reference database | Preferred over GreenGenes for detecting low-abundance rumen microbiota [7] |
| ZymoBIOMICS Mock Communities | Extraction and sequencing controls | Validate detection thresholds and quantify technical variance [100] |
| ChronoStrain | Bayesian model for strain-level profiling | Optimized for longitudinal tracking of low-abundance strains [101] |
| MetaPhlAn 4 | Taxonomic profiler with expanded database | Integrates MAGs to detect previously uncharacterized taxa [102] |
| QIIME & mothur | Standard 16S analysis pipelines | Run in parallel with SILVA database for comparative sensitivity analysis [7] |
Benchmarking QIIME and Mothur: Insights from Comparative Studies
Within the framework of a broader thesis on bioinformatics pipelines for 16S rRNA data analysis, selecting the appropriate software and reference database is a critical foundational step. The choice between popular tools like QIIME and mothur can systematically influence the taxonomic profile obtained, particularly affecting the ecological interpretation of microbial communities [8]. This application note provides a structured, evidence-based comparison of these two pipelines, focusing specifically on their differential agreement in classifying abundant versus rare taxa—a key consideration for researchers, scientists, and drug development professionals aiming to derive robust biological insights from their data.
Quantitative Comparison of Pipeline Performance
A direct comparison of QIIME and mothur, using both the GreenGenes (GG) and SILVA reference databases on rumen microbiota samples from dairy cows, revealed that while overall results are comparable, critical differences emerge at different levels of taxonomic abundance [8].
Table 1: Comparison of Genera Assignment Using GreenGenes Database
| Metric | QIIME | Mothur | Common Genera |
|---|---|---|---|
| Total Genera Assigned | 24 | 29 | 23 |
| Avg. RA of Tool-Exclusive Genera | 0.19% | 2.89% (SD=9.67) | - |
| Avg. RA of Shared Genera | - | - | 2.60% (SD=8.30) |
| Unassigned OTUs to Genus | 61% (SD=2.7) | 67% (SD=2.5) | - |
Table 2: Comparison of Genera Assignment Using SILVA Database
| Metric | QIIME | Mothur | Common Genera |
|---|---|---|---|
| Total Genera Assigned | 13 | 3 | 52 |
| Avg. RA of Tool-Exclusive Genera | 0.28% (SD=0.13) | 1.90% (SD=6.51) | - |
| Avg. RA of Shared Genera | - | - | 1.79% (SD=5.67) |
- Agreement on Abundant Taxa: For highly abundant genera (Relative Abundance, RA > 1%), such as Prevotella and Succiniclasticum, both pipelines showed a high degree of agreement, with no statistical differences in their overall relative abundance estimates, regardless of the reference database used [8].
- Divergence on Rare Taxa: Significant differences ((P < 0.05)) were observed for less common genera (RA < 10%). When using the GreenGenes database, mothur consistently assigned OTUs to a larger number of genera and assigned higher relative abundances to these rare microorganisms compared to QIIME [8].
- Impact on Diversity Metrics: These discrepancies directly influenced alpha and beta diversity measures. Mothur reported significantly larger richness and more favorable rarefaction curves, indicating higher sensitivity for low-abundance taxa. This led to significant differences in the calculated dissimilarity between samples (beta diversity), which was attenuated but not eliminated when using the SILVA database [8].
Experimental Protocols for Pipeline Comparison
The following detailed methodology outlines the key steps for a head-to-head comparison of QIIME and mothur, as derived from the cited literature [8].
Sample Preparation and Sequencing
- Sample Source: Rumen content samples were collected from a cohort of 18 dairy cows.
- Library Preparation: The hypervariable V4 region of the 16S rRNA gene was amplified using the Nextera kit for library preparation.
- Sequencing: 250 bp paired-end sequencing was performed on an Illumina MiSeq platform.
Bioinformatic Analysis with Two Pipelines
- Software Versions: QIIME package version 1.9.1 and mothur version 1.39.5 were used.
- Reference Databases: Both pipelines were run using the GreenGenes (May 2013 version) and SILVA (release 132) databases for taxonomic assignment.
- Data Processing: After filtering and chimera removal, the average number of sequences per sample was 54,544 (SD=9,041) for QIIME and 53,790 (SD=7,709) for mothur, which was not a statistically significant difference. These sequences were then clustered into Operational Taxonomic Units (OTUs) according to each pipeline's default workflow.
Assessment Framework for Qualitative and Quantitative Evaluation
An independent assessment framework utilizing titrated mixtures of environmental samples (e.g., human stool DNA) can be employed to evaluate the qualitative and quantitative characteristics of the count tables generated by different pipelines [104].
- Qualitative Assessment (Feature Presence/Absence): This evaluation estimates the proportion of artifactual features (false positives) in a count table by identifying features that are only present in unmixed samples or specific titrations and cannot be accounted for by random sampling alone. When combined with sparsity metrics (the proportion of zero-valued cells), it helps diagnose if a pipeline has a higher false-positive or false-negative rate [104].
- Quantitative Assessment (Feature Abundance): This evaluation measures the agreement between the observed relative abundance (and differential abundance) values in the mixtures and the expected values calculated from the unmixed samples and the mixture design. It assesses the bias and variance of a pipeline's abundance estimates [104].
Workflow and Relationship Diagrams
Figure 1: Bioinformatics workflow for comparing QIIME and mothur pipelines, showing divergent outcomes for abundant versus rare taxa.
Figure 2: Logical relationship showing how database choice critically influences pipeline agreement on rare taxa and downstream ecological analysis.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for 16S rRNA Pipeline Comparisons
| Item | Function in the Experiment |
|---|---|
| Illumina MiSeq Platform | High-throughput sequencing platform for generating 250 bp paired-end 16S rRNA sequence data. |
| Nextera Kit | Used for library preparation and amplification of the target hypervariable region (e.g., V4). |
| GreenGenes Database | A reference database for taxonomic assignment; shown to produce greater divergence for rare taxa. |
| SILVA Database | A curated reference database for taxonomic assignment; recommended for improved agreement between pipelines. |
| QIIME Software | A comprehensive bioinformatics pipeline suite for processing and analyzing 16S rRNA sequencing data. |
| Mothur Software | A bioinformatics pipeline suite for analyzing 16S rRNA sequence data, with high sensitivity for rare taxa. |
In the field of microbial ecology, the analysis of 16S rRNA gene sequencing data relies heavily on bioinformatic pipelines to translate raw sequence data into meaningful biological insights. The choice of pipeline is a critical methodological decision that directly influences the estimation of key diversity metrics, including richness (alpha-diversity) and between-sample diversity (beta-diversity). This application note examines the specific effects of choosing between two prevalent platforms—QIIME and mothur—on these metrics, providing structured experimental data and protocols to guide researchers in making informed, reproducible choices for their 16S rRNA analyses. The evidence presented herein is framed within a broader thesis on bioinformatics pipeline selection, underscoring that the tool chosen is not a neutral facilitator but an active determinant of the resulting microbial community structure [7] [8] [17].
Comparative Analysis of Pipeline Performance on Diversity Metrics
Key Findings on Richness and Beta-Diversity
A direct comparison of QIIME (v1.9.1) and mothur (v1.39.5) using 16S rRNA amplicon sequences from rumen microbiota demonstrated that the software choice significantly impacts the observed diversity, particularly for low-abundance taxa [7] [8].
Table 1: Impact of Bioinformatics Pipeline on Taxonomic Richness
| Reference Database | Software | Average Number of Genera Detected | Statistical Significance (Richness) |
|---|---|---|---|
| GreenGenes | QIIME | 24 | P < 0.05 |
| GreenGenes | mothur | 29 | P < 0.05 |
| SILVA | QIIME | 65 (13 exclusive) | Not Significant |
| SILVA | mothur | 55 (3 exclusive) | Not Significant |
Table 2: Impact on Beta-Diversity Analysis
| Factor | Impact on Beta-Diversity (Between-Sample Dissimilarity) |
|---|---|
| Software Choice (with GreenGenes) | Significant and relevant differences in identified dissimilarity between pairs of samples. |
| Software Choice (with SILVA) | Differences were attenuated, but not erased. |
| Database Choice | SILVA database reduced the inter-pipeline discrepancy in beta-diversity metrics. |
The analysis revealed that mothur consistently clustered sequences into a larger number of OTUs across both databases, which translated into higher observed richness before the application of abundance filters [7] [8]. This effect was more pronounced when using the GreenGenes database. Furthermore, these differences in OTU clustering and assignment led to significant differences in beta-diversity estimates, meaning the perceived dissimilarity between microbial communities varied depending on the pipeline used [8].
The Role of the Reference Database
The choice of reference database (GreenGenes vs. SILVA) interacts with the software choice. The aforementioned differences, particularly for low-abundance taxa and beta-diversity, were markedly reduced when the SILVA database was used for taxonomic classification [7] [8]. This suggests that SILVA may be a preferred reference dataset for certain environments, like the rumen, as it promotes greater consistency between QIIME and mothur outputs.
Detailed Experimental Protocol for Pipeline Comparison
To ensure the reproducibility of the findings summarized in this note, the following detailed protocol outlines the key steps used in the comparative study.
Sample Processing and Sequencing
- Sample Source: Rumen content was collected from a cohort of 18 dairy cows [8].
- Library Preparation: The hypervariable V4 region of the 16S rRNA gene was amplified. Libraries were constructed using the Nextera kit (Illumina) [7] [8].
- Sequencing: 250 bp paired-end sequencing was performed on a MiSeq platform (Illumina) [7] [8].
Bioinformatics Analysis with QIIME and mothur
The following workflow diagrams and steps detail the parallel processing of sequences through the two pipelines.
QIIME Protocol (v1.9.1)
- Demultiplexing and Quality Filtering: Use
split_libraries.pyto assign multiplexed reads to samples based on their barcodes and perform quality filtering. Default parameters can include a minimum quality score of 25 and removal of ambiguous base calls [105]. - OTU Clustering: Cluster quality-filtered sequences into Operational Taxonomic Units (OTUs) using a method such as
uclust[17]. - Taxonomic Assignment: Assign taxonomy to representative sequences from each OTU by aligning them against a reference database (e.g., GreenGenes or SILVA) [7] [8].
- Diversity Analysis: Calculate alpha-rarefaction curves and generate beta-diversity distance matrices (e.g., using weighted/unweighted Unifrac) from the resulting OTU table [105].
- Demultiplexing and Quality Filtering: Use
mothur Protocol (v1.39.5)
- Assembly and Quality Control: Use
make.contigsto combine paired-end reads. Subsequently, perform alignment to a reference alignment (e.g., SILVA) withalign.seqs, followed by rigorous quality screening (screen.seqs) and filtering (filter.seqs) to remove poorly aligned regions and gaps [43]. - OTU Clustering: Calculate pairwise distances between sequences using
dist.seqsand then cluster them into OTUs with theclustercommand [43] [106]. - Taxonomic Classification: Classify sequences against a reference taxonomy (e.g., RDP, SILVA, or GreenGenes) using the
classify.otucommand [43]. - Diversity Analysis: Generate rarefaction curves and calculate similarity matrices between samples to analyze within- and between-sample diversity [107].
- Assembly and Quality Control: Use
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Materials and Reagents for 16S rRNA Pipeline Analysis
| Item | Function / Role in Analysis | Example / Note |
|---|---|---|
| Silva SSU Reference Database | A curated, high-quality alignment used for sequence alignment and taxonomic classification; promotes consistency between pipelines. | Release 132 was used in the cited study [7] [8]. |
| GreenGenes Database | A popular, yet now static, 16S rRNA gene database for taxonomic classification. | May lead to larger inter-pipeline differences compared to SILVA [7] [8]. |
| Mock Microbial Community | A controlled mixture of known microbial strains used to benchmark pipeline accuracy, error rates, and sensitivity. | Essential for validating and optimizing any new workflow [43] [17]. |
| Illumina MiSeq Platform | A high-throughput sequencing platform capable of generating 250 bp paired-end reads for 16S rRNA amplicons. | The standard platform for studies of this kind [7] [43]. |
| Nextera XT DNA Library Prep Kit | Used for preparing sequencing libraries, including tagmentation and indexing of amplicons. | Enables multiplexing of numerous samples in a single sequencing run [7] [8]. |
The evidence demonstrates conclusively that the choice of bioinformatics pipeline (QIIME vs. mothur) is a significant source of variation in 16S rRNA analysis, directly impacting fundamental diversity metrics like richness and beta-diversity. To ensure robust and reproducible results, researchers should:
- Standardize the Pipeline: Once a pipeline and database combination is selected, use it consistently throughout a study to enable valid comparisons.
- Use the SILVA Database: Consider using the SILVA database for taxonomic classification, as it has been shown to attenuate differences between QIIME and mothur outputs [7] [8].
- Report Methodology in Detail: Always explicitly report the software versions, reference databases, and key parameters used in bioinformatic analyses to ensure transparency and reproducibility.
- Validate with Mock Communities: Where possible, include mock community samples to benchmark the performance and error profiles of the chosen pipeline [17].
Within the framework of a broader thesis on bioinformatics pipelines for 16S rRNA data analysis, the selection of a reference database is a critical methodological step that profoundly influences downstream biological interpretations. Taxonomic identification is a cornerstone of microbial ecology, and in amplicon-based metagenomic studies, this process is inherently tied to the reference database used for sequence classification [70]. Among the most widely used resources in pipelines like QIIME and mothur are the SILVA and Greengenes databases. Each database possesses a unique set of characteristics regarding curation methodology, taxonomic scope, and update frequency, which collectively contribute to observable differences in the resulting taxonomic profiles. This application note delineates the specific effects of database choice on taxonomic assignment within QIIME and mothur environments. It provides structured comparisons and detailed protocols to guide researchers, scientists, and drug development professionals in making informed decisions that enhance the reproducibility and reliability of their microbiome studies.
Database Fundamentals and Comparative Characteristics
Core Architectural Philosophies
The SILVA and Greengenes databases were constructed with different primary objectives and curation philosophies, which underpin their performance variations.
SILVA Database: The SILVA project (from Latin silva, forest) provides a comprehensive, quality-checked resource for aligned ribosomal RNA gene sequences from all three domains of life (Bacteria, Archaea, and Eukarya) [71] [72]. A key feature of SILVA is its semi-automatic data curation procedure, which integrates information from authoritative resources including the Genome Taxonomy Database (GTDB), the List of Prokaryotic names with Standing in Nomenclature (LPSN), and Bergey's Manual of Systematic Bacteriology [73]. The taxonomy is manually curated and follows a defined priority rule for cultured prokaryotes: Bergey's > GTDB > LPSN > Users > NCBI [73]. SILVA databases are released periodically, with a strong focus on providing a phylogenetic framework based on guide trees. The project also includes a unique feature of incorporating 'Candidatus' taxa and names without standing in nomenclature [73].
Greengenes Database: Greengenes is a 16S rRNA gene database specifically for Bacteria and Archaea, historically notable for being the default database in the widely used QIIME pipeline [70] [75]. A distinguishing feature of its original construction was its comprehensive chimera screening process using the Bellerophon algorithm, which identified putative chimeras in 3% of environmental sequences and 0.2% of records derived from isolates [75]. However, a critical limitation of the original Greengenes database is that its last release was in August 2013, meaning it does not incorporate the vast number of novel bacterial sequences discovered since then [70] [76]. It is important to note a next-generation database, Greengenes2, has been introduced to address this gap, leveraging a phylogeny backed by whole genomes and integrating with the GTDB taxonomy [77].
Quantitative Database Comparison
The table below summarizes the key differentiating characteristics of the classic SILVA and Greengenes databases as referenced in the literature.
Table 1: Key Characteristics of SILVA and Greengenes Databases
| Characteristic | SILVA | Greengenes (classic) |
|---|---|---|
| Domain Coverage | Bacteria, Archaea, Eukarya [71] [72] | Bacteria & Archaea only [70] [75] |
| Update Status | Regularly updated (e.g., releases 138, 138.2) [71] | Not updated since August 2013 [70] [76] |
| Number of SSU sequences | ~190,000 (Release 111) [70] | ~99,000 (Release 13_8) [70] |
| Species-Level Annotations | Available but can be incomplete; some entries have only strain information [70] | Limited; ~10% of sequences have species-level names [70] [76] |
| Primary Use Case | Broad-range taxonomy assignment across all life domains; preferred for full-length and V3-V4 analyses [108] [15] | Default for older QIIME versions; primarily for 16S rRNA gene analysis [70] [109] |
| Key Curation Feature | Integration with GTDB & LPSN; manual curation based on guide trees [73] | Chimera-checked; standard alignment; multiple taxonomies tracked [75] |
Impact on Taxonomic Profiling and Diversity Metrics
Mock Community Evaluations and Accuracy
Studies using mock microbial communities, where the true composition is known, provide the most direct evidence of database-driven discrepancies in taxonomic assignment accuracy.
Genus and Species-Level Identification: A comparative evaluation using public mock community data (PRJEB6244) revealed significant performance differences. At the genus level, the EzBioCloud database identified over 40 true positive genera out of 44 present, while Greengenes found only about 30, and SILVA, though finding a sufficient number of genera, had the highest rate of false-positives (around 20% of predicted genera were incorrect) [70]. The differences were more pronounced at the species level. EzBioCloud correctly identified about 40 species, whereas SILVA identified far fewer correct species, and Greengenes found only a few [70]. This performance gap is largely attributed to the fact that Greengenes and SILVA contain sequences with missing or incomplete species-level taxonomic information [70].
Underlying Causes of Discrepancy: The higher number of false-positive assignments observed with SILVA is partly a function of its larger size (~190,000 sequences at the time of the study versus ~99,000 in Greengenes), which increases the probability of a sequence being incorrectly assigned to a different genus [70]. Furthermore, the outdated nature of the classic Greengenes database means it lacks many novel bacterial sequences and updated taxonomic reclassifications, leading to an under-detection of known genera and a higher false-negative rate [70].
In-Sample Diversity Estimation
The choice of reference database also significantly impacts the calculation of alpha diversity indices, which are crucial for estimating microbial richness and evenness within a sample.
Richness and Evenness Indices: Analysis of a uniformly distributed mock community demonstrated that the database choice systematically affects alpha diversity metrics. When using the same clustering method, SILVA tended to overestimate sample richness (Observed and Chao1 indices), while Greengenes also overestimated richness and underestimated evenness (Simpson’s index) compared to the known truth [70]. In contrast, the EzBioCloud database provided richness estimates closer to the true value and higher, more accurate Simpson's evenness values [70].
Biological Interpretation: These findings indicate that using SILVA or Greengenes can lead to a perception of a community that is richer and less even than it truly is, which could directly affect ecological conclusions drawn from the data. The overestimation of richness is likely linked to the higher number of sequences in these databases, which, if not perfectly curated, can cause single species to be split into multiple operational taxonomic units (OTUs) due to sequencing errors, thereby inflating richness counts [70].
Real-World Study Comparisons
The influence of the database extends beyond controlled mock communities into real-world research scenarios, such as the analysis of complex microbial environments.
- Rumen Microbiota Study: A comparative study of rumen microbiota composition found that while both QIIME and mothur pipelines identified the same most abundant genera (e.g., Prevotella, Butyrivibrio) regardless of the database, significant differences emerged for less abundant taxa (relative abundance < 10%) [15]. When using Greengenes, mothur assigned OTUs to a larger number of genera and assigned higher relative abundances for these less frequent microorganisms compared to QIIME, resulting in mothur reporting significantly larger richness [15]. These differences in low-abundance taxa led to relevant discrepancies in beta diversity (dissimilarity between samples) between the pipelines. However, these differences were attenuated when the SILVA database was used as the reference, with both pipelines producing more comparable richness, diversity, and relative abundances for common rumen microbes [15]. This led the authors to suggest that SILVA is a preferred reference dataset for classifying OTUs from rumen microbiota [15].
Practical Protocols for Database Selection and Application
Decision Workflow for Database and Pipeline Selection
The following diagram outlines a logical workflow to guide researchers in selecting an appropriate reference database and analysis pipeline based on their specific experimental context.
Diagram 1: A workflow for selecting 16S rRNA reference databases and pipelines.
Detailed Experimental Protocol for Database Comparison
For researchers aiming to validate the impact of database choice on their specific dataset, the following comparative protocol is recommended.
Table 2: Research Reagent and Computational Solutions
| Item Name | Function/Description | Example Source/Format |
|---|---|---|
| QIIME 2 | A powerful, extensible, and decentralized microbiome analysis platform with a focus on data and analysis transparency. | https://qiime2.org/ |
| mothur | An open-source, expandable software pipeline for microbiome data, encompassing all traditional 16S rRNA analysis steps. | https://mothur.org/ |
| SILVA SSU Ref NR 99 | A high-quality, non-redundant dataset of aligned small subunit (SSU) ribosomal RNA sequences for reference-based classification. | https://www.arb-silva.de/ (QIIME-compatible format) |
| Greengenes2 | A modern 16S rRNA database redesigned from whole genomes, focusing on harmonizing 16S and shotgun data. | http://ftp.microbio.me/greengenes_release/ |
| Mock Community | A control sample containing a known, defined composition of microbial strains for benchmarking and accuracy assessment. | e.g., ZymoBIOMICS, ATCC MSA-1000 |
Step-by-Step Procedure:
Data Preparation and Quality Control
- Obtain your 16S rRNA sequencing data (e.g., FASTQ files) and a publicly available mock community dataset (e.g., ENA accession PRJEB6244) [70].
- Perform initial quality control on all sequence data. In QIIME 2, this can be done using
q2-demuxfollowed byq2-dada2orq2-deblurfor denoising and generation of amplicon sequence variants (ASVs). In mothur, follow the standard operating procedure (Miseq_SOP) involvingmake.contigs,screen.seqs, andchimera.uchime.
Parallel Taxonomic Classification
- In QIIME 2: Use the
feature-classifierplugin. For V4 data with Greengenes2, utilize theqiime greengenes2 filter-featuresandqiime greengenes2 taxonomy-from-tablecommands as detailed in the forum tutorial [77]. For other regions or for using SILVA, train a classifier on the appropriate region of the SILVA database usingfit-classifier-naive-bayes. - In mothur: Use the
classify.seqscommand. Specify the reference files for each database (e.g.,reference=silva.nr_v138.alignandtaxonomy=silva.nr_v138.taxfor SILVA; similarly formatted files for Greengenes).
- In QIIME 2: Use the
Diversity Analysis and Comparison
- Generate alpha and beta diversity metrics within each pipeline using the taxonomy assignments derived from each database.
- For the mock community data, calculate accuracy metrics: True Positives (TP), False Positives (FP), and False Negatives (FN) at genus and species levels, comparing the assignments to the known composition [70].
- For your real samples, compare the relative abundance of key taxa, overall community richness (e.g., Chao1), and between-sample diversity (e.g., UniFrac distances) generated from the SILVA-based versus Greengenes-based classifications.
Data Synthesis and Reporting
- Create comparative tables and visualizations (e.g., bar plots of relative abundances, PCoA plots for beta diversity) to highlight the differences.
- Document the specific database versions and pipeline parameters used to ensure future reproducibility.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Resources
| Tool/Resource | Function in Analysis |
|---|---|
| SILVA SSU Ref NR 99 | A curated, non-redundant reference dataset for taxonomy assignment, encompassing Bacteria, Archaea, and Eukarya. Recommended for general use and cross-pipeline consistency [72] [15]. |
| Greengenes2 | A modern 16S rRNA database that integrates genome-based taxonomy. Particularly suited for V4 region studies in QIIME2 and for projects aiming to correlate 16S and shotgun metagenomic data [77]. |
| EzBioCloud Database | A database optimized for species-level identification. Consider for studies where high taxonomic resolution is paramount, as it has demonstrated high accuracy in mock community tests [70]. |
| QIIME 2 Framework | A modular, scalable analysis platform with integrated database resources and a focus on reproducibility. Ideal for standardized, high-throughput processing [108] [77]. |
| mothur Pipeline | A comprehensive, all-in-one software pipeline. Well-suited for users who prefer a single tool to conduct all analysis steps from raw sequences to community analyses [76] [15]. |
| Public Mock Community Data | A benchmark dataset with known composition (e.g., PRJEB6244) for empirically testing pipeline and database accuracy before analyzing study data [70]. |
The choice between SILVA and Greengenes is not merely a technical formality but a decisive factor that shapes the taxonomic profile of a microbial community. Evidence from mock community and real-world studies consistently shows that SILVA, being regularly updated and more comprehensive, often provides a more reliable and consistent classification, especially across different analysis pipelines [15]. In contrast, the classic Greengenes database, while historically important, is now outdated and can lead to under-detection of taxa and inflated diversity metrics [70] [76]. The emergence of Greengenes2 offers a modernized alternative, particularly for QIIME2 users working with the V4 region [77]. For drug development professionals and researchers, where accurate biological interpretation is critical, the protocol of benchmarking database choices against a mock community relevant to their study system is strongly recommended. This practice ensures that the conclusions drawn from complex bioinformatics pipelines are grounded in a clear understanding of the methodological biases introduced at the level of fundamental reference resources.
In the field of microbial ecology, 16S rRNA gene sequencing has become an indispensable method for profiling complex microbial communities across diverse environments, from the human gut to soil ecosystems [99] [7]. However, this powerful analytical approach remains vulnerable to multiple sources of technical error introduced throughout the experimental workflow, including DNA extraction biases, PCR amplification artifacts, chimeric sequence formation, and platform-specific sequencing errors [99] [110]. These errors significantly impact the accuracy of microbial composition data and subsequent biological interpretations. Without proper validation, erroneous sequences can artificially inflate diversity metrics and lead to incorrect taxonomic assignments.
Mock microbial communities—composed of genomic DNA from known bacterial strains in defined proportions—provide an essential experimental control for assessing error rates and validating bioinformatics pipelines [99] [110]. By comparing sequencing results against the expected composition of these mocks, researchers can quantify the error rate of their entire workflow, from sample preparation to data analysis. Recent benchmarking studies utilizing complex mock communities have revealed substantial differences in performance between popular analysis tools like QIIME and mothur, as well as between different algorithmic approaches for defining taxonomic units [99] [7]. These findings underscore the critical importance of mock community validation in ensuring the reliability of 16S rRNA sequencing data, particularly for clinical and pharmaceutical applications where accurate microbial identification can inform therapeutic development.
Comparative Performance of Bioinformatics Tools
OTU vs. ASV Approaches: A Benchmarking Analysis
The analysis of 16S rRNA sequencing data primarily employs two methodological approaches: Operational Taxonomic Units (OTUs) clustered at a fixed similarity threshold (typically 97%), and Amplicon Sequence Variants (ASVs) generated through denoising algorithms that attempt to distinguish biological sequences from technical errors [99]. A comprehensive benchmarking study utilizing the most complex mock community to date (227 bacterial strains across 197 species) revealed distinct performance characteristics between these approaches [99].
ASV algorithms, particularly DADA2, demonstrated highly consistent output but were prone to over-splitting single biological sequences into multiple variants. This over-splitting likely results from the inability of these algorithms to fully account for intragenomic variation between multiple 16S rRNA gene copies within the same organism [99] [33]. Conversely, OTU-based methods like UPARSE produced clusters with lower error rates but exhibited more over-merging of biologically distinct sequences into single units. Notably, both UPARSE and DADA2 showed the closest resemblance to the expected microbial composition, particularly for alpha and beta diversity metrics [99].
Table 1: Performance Comparison of OTU and ASV Algorithms Using Mock Communities
| Algorithm | Type | Error Rate | Tendency | Similarity to Expected Composition |
|---|---|---|---|---|
| DADA2 | ASV | Low | Over-splitting | High |
| UPARSE | OTU | Low | Over-merging | High |
| Deblur | ASV | Moderate | Over-splitting | Moderate |
| MED | ASV | Moderate | Over-splitting | Moderate |
| UNOISE3 | ASV | Moderate | Over-splitting | Moderate |
| Opticlust | OTU | Moderate | Over-merging | Moderate |
QIIME vs. Mothur: Database-Dependent Discrepancies
The choice of bioinformatics pipeline significantly impacts taxonomic assignment and diversity estimates. A comparative study of rumen microbiota revealed that while QIIME and mothur show strong agreement for abundant genera (RA > 1%), notable differences emerge for less abundant taxa [7]. Mothur consistently identified a larger number of OTUs and microbial genera, particularly when using the GreenGenes database, resulting in richer observed communities compared to QIIME [7].
These differences were especially pronounced for low-abundance microorganisms (RA < 10%), where mothur assigned sequences to a larger number of genera at higher relative abundances. This discrepancy substantially influenced beta diversity measurements between samples, suggesting that the choice of analysis pipeline can affect the perceived dissimilarity between microbial communities [7]. The database selection proved crucial, with SILVA producing more comparable results between pipelines than GreenGenes, making it the preferred reference database for minimizing inter-pipeline variability [7].
Table 2: QIIME vs. Mothur Performance with Different Reference Databases
| Metric | Tool | GreenGenes Database | SILVA Database |
|---|---|---|---|
| Number of OTUs | QIIME | Lower | Moderate |
| Mothur | Higher | Moderate | |
| Genera Assigned (RA > 0.1%) | QIIME | 24 | Moderate |
| Mothur | 29 | Moderate | |
| Unassigned OTUs | QIIME | 61% | Improved |
| Mothur | 67% | Improved | |
| Analytical Sensitivity | QIIME | Lower | Moderate |
| Mothur | Higher | Moderate |
Emerging Tools: Kraken 2 and Bracken
Recent advancements in computational methods have introduced alternative approaches for 16S rRNA data analysis. Kraken 2 with Bracken demonstrates exceptional speed and accuracy for 16S rRNA profiling, achieving up to 300 times faster processing with 100-fold less RAM usage compared to QIIME 2's q2-feature-classifier while generating more accurate community profiles [98]. This combination provides a particularly efficient solution for large-scale studies where computational resources may be limiting.
Experimental Factors Influencing Accuracy
Primer Selection and Target Region
The choice of 16S rRNA variable region significantly impacts taxonomic resolution and community composition results. Different hypervariable regions exhibit substantial variation in their ability to discriminate between bacterial taxa [110] [33]. The V4 region, despite its popularity, performs poorest for species-level discrimination, failing to confidently classify 56% of sequences in in-silico experiments [33]. In contrast, the V1-V2 and V3-V5 regions provide better taxonomic resolution, though with taxon-specific biases [110].
Different primer sets also exhibit platform-specific performance characteristics. Studies comparing Illumina MiSeq and Ion Torrent PGM platforms found that Ion PGM primers detected more expected mock community species than their MiSeq counterparts, though the V4-V5 primers showed the most consistent results across platforms [110]. The targeting of multiple regions, as implemented in the Ion Torrent 16S Metagenomics Kit (which amplifies V2-4-8 and V3-6,7-9), presents analytical challenges but may provide complementary information [111].
Table 3: Performance of Different 16S rRNA Gene Regions Based on In-Silico Analysis
| Target Region | Species-Level Classification Efficiency | Taxonomic Biases | Recommended Applications |
|---|---|---|---|
| V4 | Lowest (44% classified) | Minimal taxon-specific bias | General diversity studies |
| V1-V2 | Moderate (60% classified) | Poor for Proteobacteria | Specific taxon-focused studies |
| V3-V5 | Moderate (58% classified) | Poor for Actinobacteria | Broad-range detection |
| V1-V3 | High (65% classified) | Moderate across taxa | General purpose |
| V6-V9 | Variable | Best for Clostridium and Staphylococcus | Targeted studies |
| Full-length (V1-V9) | Highest (95% classified) | Minimal biases | Maximum resolution |
Sequencing Platform Considerations
The choice of sequencing platform introduces distinct error profiles that must be accounted for during mock community validation. Illumina platforms primarily exhibit nucleotide substitution errors, while Ion Torrent shows higher rates of indel errors, particularly in homopolymer regions [110] [33]. Full-length 16S rRNA sequencing using PacBio circular consensus sequencing (CCS) can achieve error rates below 1.0% with sufficient passes (≥10), enabling discrimination of single-nucleotide differences between intragenomic 16S gene copies [33].
Recent advances in third-generation sequencing technologies have made full-length 16S gene sequencing increasingly accessible, providing superior taxonomic resolution compared to short-read platforms targeting sub-regions [33]. However, the analysis of full-length 16S sequences must account for intragenomic variation between multiple 16S rRNA gene copies within a single organism, which can be misinterpreted as distinct taxa if not properly handled [33].
Standardized Protocols for Mock Community Validation
Sample Preparation and Sequencing
Mock Community Selection: Begin with commercially available mock communities (e.g., HM-782D or HC227) or custom-designed mixtures of known bacterial strains. Complex mocks comprising 200+ strains provide the most rigorous validation [99] [110].
DNA Extraction: Employ standardized extraction protocols across all samples. The QIAamp DNA Stool Mini Kit and repeat bead beating (RBB) method yield comparable results for mock communities [110].
Library Preparation: Amplify the V4-V5 region using primers 5'-CCTACGGGNGGCWGCAG-3' and 5'-GACTACHVGGGTATCTAATC-3' for Illumina platforms [99]. Include negative controls to detect contamination and positive controls (mock community) in each sequencing run.
Sequencing Parameters: Utilize paired-end sequencing (2 × 300 bp for Illumina MiSeq) to ensure sufficient overlap for error correction. Subsampling to 30,000 reads per sample provides a reasonable balance between depth and computational requirements [99].
Bioinformatics Processing
The following workflow implements a standardized approach for mock community analysis using either QIIME or mothur:
Quality Control and Preprocessing:
- Assess sequence quality using FastQC (v.0.11.9)
- Remove primer sequences using cutPrimers (v2.0) [99]
- Merge paired-end reads using USEARCH fastq_mergepairs or DADA2's read merging
- Filter sequences with ambiguous characters or high error rates (maxee_rate = 0.01)
Denoising/Clustering Implementation: For QIIME2 with DADA2:
For Mothur with UPARSE:
Taxonomic Classification:
- Align sequences to reference databases (SILVA v132 or Greengenes) [7]
- Apply the RDP classifier with a confidence threshold of 0.8
- For full-length sequences, utilize the GreenGenes full-length classifier [111]
Error Rate Calculation and Validation Metrics
Error Rate Quantification: Calculate the overall error rate as: [ \text{Error Rate} = \frac{\text{Number of erroneous sequences}}{\text{Total sequences}} \times 100] where erroneous sequences include chimeras, misclassified taxa, and sequences from contaminants not present in the mock community.
Community Composition Accuracy:
- Compare observed vs. expected relative abundances using Bray-Curtis dissimilarity
- Calculate precision and recall for taxon detection: Precision = True Positives / (True Positives + False Positives) Recall = True Positives / (True Positives + False Negatives)
Diversity Metric Validation:
- Compare observed vs. expected alpha diversity (Shannon, Faith's PD)
- Assess beta diversity preservation using weighted/unweighted UniFrac distances [111]
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents and Computational Tools for Mock Community Validation
| Category | Specific Product/Software | Function in Validation |
|---|---|---|
| Mock Communities | HM-782D (BEI Resources) | Known composition reference standard |
| HC227 (227 strain community) | Complex validation standard [99] | |
| DNA Extraction Kits | QIAamp DNA Stool Mini Kit | Standardized DNA isolation |
| Repeat Bead Beating Method | Mechanical lysis protocol [110] | |
| Sequencing Kits | Illumina MiSeq Reagent Kit v3 (600-cycle) | V4-V5 region sequencing |
| Ion Torrent 16S Metagenomics Kit | Multi-region amplification [111] | |
| Bioinformatics Tools | QIIME 2 (q2-feature-classifier) | Integrated analysis pipeline [98] |
| Mothur (Opticlust, dist.seqs) | OTU-based clustering [7] | |
| DADA2 (denoise-single/paired) | ASV generation [99] | |
| Kraken 2 + Bracken | Rapid taxonomic classification [98] | |
| Reference Databases | SILVA (release 132) | Curated alignment and taxonomy [7] |
| Greengenes (13_8) | 16S rRNA gene database [98] | |
| RDP (11.5) | Ribosomal Database Project [98] |
Mock community validation represents an essential component of rigorous 16S rRNA sequencing studies, particularly in pharmaceutical and clinical research where accurate microbial identification is critical. Based on current benchmarking studies, the following best practices are recommended:
Implement Mock Communities in Every Sequencing Run: Include mock community controls in each batch to monitor technical variability and platform performance over time.
Select Appropriate Bioinformatics Tools: Consider the trade-offs between ASV and OTU approaches—ASV methods like DADA2 provide higher resolution but may over-split genuine biological sequences, while OTU methods like UPARSE offer more robust clustering at the potential cost of merging distinct taxa [99].
Utilize the SILVA Database: Standardize taxonomic classification using the SILVA database, which produces more consistent results between QIIME and mothur pipelines compared to GreenGenes [7].
Target Appropriate Variable Regions: When using short-read platforms, select variable regions that provide sufficient taxonomic resolution for your research question—V1-V3 or V3-V5 regions generally outperform V4 alone [33].
Validate Full-Length Sequencing: For PacBio or Nanopore platforms, establish specific validation protocols that account for intragenomic 16S copy variation and platform-specific error profiles [33].
Report Validation Metrics: Transparently document error rates, precision, recall, and diversity preservation metrics from mock community analysis to establish the reliability of experimental results.
As sequencing technologies continue to evolve, mock community validation remains the gold standard for ensuring the accuracy and reproducibility of 16S rRNA-based microbial community analyses in drug development and clinical research.
For years, 16S rRNA gene (DNA-based) sequencing has been the cornerstone of microbial ecology, enabling the taxonomic census of bacterial communities across diverse environments. However, this approach captures all DNA present in a sample, including that from dead, dormant, or transient cells, as well as extracellular DNA persisting in the environment [112] [113]. This fundamental limitation can obscure the true picture of metabolically active community members driving ecological processes. The analysis of 16S rRNA transcripts (RNA-based) has emerged as a powerful complementary approach that reveals the active subset of a microbial community by targeting the ribosomes present in living cells [113]. This application note compares DNA- and RNA-based approaches for identifying active communities, providing structured experimental protocols, and contextualizing these methods within modern bioinformatics pipelines such as QIIME and mothur.
Key Conceptual Differences Between DNA and RNA Approaches
The choice between DNA and RNA-based 16S analysis depends on the specific research question, each offering distinct advantages and limitations as summarized in the table below.
Table 1: Fundamental comparison of DNA-based versus RNA-based 16S analysis
| Feature | 16S rRNA Gene (DNA-based) | 16S rRNA Transcript (RNA-based) |
|---|---|---|
| Target Molecule | Genomic DNA (gene copies) | RNA transcripts (ribosomal RNA) |
| Biological Meaning | Total microbial community membership (living, dead, dormant) | Potentially active community (proxy for protein synthesis potential) |
| Sensitivity | Lower (1-21 gene copies per cell [112]) | Higher (e.g., ~25,000 ribosomes per E. coli cell [114]) |
| Technical Bias | rRNA gene copy number variation between taxa [112] | Ribosome number per cell, influenced by growth rate and cell size [112] |
| Stability of Target | Highly stable | Rapidly degraded |
| Information Gained | Community structure and taxonomic composition | Active portion of the community, often revealing different ecological drivers [113] |
Comparative Performance Data
Recent studies across different ecosystems have quantitatively compared the outcomes of DNA and RNA co-analysis, consistently demonstrating that the two approaches yield distinct but complementary results.
Table 2: Empirical findings from comparative studies of DNA and RNA-based 16S analysis
| Study System | Key Finding | Reported Quantitative Differences |
|---|---|---|
| Equine Uterine Microbiome [112] [114] | RNA-based approach showed significantly higher sensitivity and detected a more diverse community. | - 10-fold higher sensitivity for RNA [112]- Significant differences in alpha (Simpson, Chao1) and beta diversity [112]- Higher number of Amplicon Sequence Variants (ASVs) and taxonomic units with RNA [112] |
| Soil Rhizosphere [113] | RNA profiles revealed fine-scale differences in genera between rhizosphere and bulk soil not apparent with DNA. | - DNA disproportionately increased the perceived importance of Saccharibacteria and Gemmatimonadetes [113]- RNA elevated the detected activity of known root associates (e.g., Comamonadaceae, Rhizobacter) [113] |
| General Implication | DNA-based community composition may not fully capture community activity, impacting ecological interpretation. | Differential abundance analysis revealed significant differences between DNA and RNA samples at all taxonomic levels [112]. |
Integrated Experimental Protocol
This section provides a detailed methodology for the simultaneous extraction and subsequent 16S amplicon sequencing of both DNA and RNA from the same sample, adapted from established protocols [112] [114] [113].
Sample Collection and Preservation
- Collection: Collect biomass using a method appropriate for the ecosystem (e.g., cytobrush for uterine samples [112], soil corer for rhizosphere studies [113]).
- Critical Step: Immediately place the sample in a cryotube containing an appropriate lysis buffer that stabilizes both DNA and RNA (e.g., RLT Plus buffer with DTT [112]).
- Storage: Flash-freeze the samples in liquid nitrogen and store at -80°C until nucleic acid extraction to preserve RNA integrity.
Simultaneous DNA and RNA Extraction
- Lysis: Add additional lysis buffer to the sample and shake vigorously (e.g., 1500 rpm for 10 min at room temperature) to ensure complete homogenization [112].
- Co-extraction: Use a commercial kit designed for the simultaneous purification of genomic DNA and total RNA (e.g., AllPrep DNA/RNA/miRNA Universal Kit from Qiagen) following the manufacturer's "Simultaneous Purification" protocol [112].
- Elution: Elute RNA and DNA in separate, nuclease-free tubes using the provided elution buffers or RNase-free water.
- Quality Control: Essential steps include:
- Measure concentration and purity with a spectrophotometer (e.g., NanoDrop).
- Assess RNA integrity using an instrument like the Agilent 2100 Bioanalyzer with the RNA 6000 Nano assay [112].
cDNA Synthesis from RNA
- DNase Treatment: Treat the purified RNA with DNase I to remove any contaminating genomic DNA.
- Reverse Transcription: Use a reverse transcriptase enzyme (e.g., SuperScript IV) and a universal reverse primer (e.g., Pro805R) or random hexamers to generate cDNA.
- Control: Always include a no-reverse-transcriptase (-RT) control to confirm the absence of DNA contamination in subsequent PCR steps.
16S rRNA Gene Amplicon PCR and Sequencing
- DNA Template: Use the extracted genomic DNA.
- RNA-derived Template: Use the synthesized cDNA.
- PCR Amplification: Amplify the hypervariable regions (e.g., V3-V4) using tailed primers (e.g., Pro341F and Pro805R) [112].
- Blocking (if needed): For samples with high host mitochondrial DNA, use a Peptide Nucleic Acid (PNA) clamp or blocking oligonucleotides to suppress amplification of host 12S rRNA [112].
- Library Preparation & Sequencing: Follow standard Illumina MiSeq protocols for dual-indexed amplicon sequencing.
The following workflow diagram illustrates the parallel processing of DNA and RNA from a single sample:
Bioinformatics Pipeline Integration
The data generated from both DNA and RNA workflows require robust bioinformatics processing. The choice of pipeline and reference database can significantly impact results, especially for low-abundance taxa.
Pipeline and Database Selection
Table 3: Comparison of bioinformatics tools and databases for 16S analysis
| Component | Option | Considerations for DNA/RNA Studies |
|---|---|---|
| Bioinformatics Pipeline | QIIME 2 [17] | User-friendly, modular, integrates Deblur (ASV) and DADA2. Strong community support. |
| mothur [7] [8] | Slightly steeper learning curve, often clusters more OTUs, especially with GreenGenes [7]. | |
| DADA2 [17] | ASV-based; offers high sensitivity but may require careful parameter tuning to balance specificity [17]. | |
| USEARCH-UNOISE3 [17] | ASV-based; provides an excellent balance between resolution and specificity [17]. | |
| Reference Database | SILVA [7] [8] | Preferred for non-human microbiomes (e.g., rumen); produces more comparable results between QIIME and mothur [7] [8]. |
| GreenGenes | May lead to larger differences in assigned OTUs and richness between pipelines [7]. |
Specific Considerations for RNA Data
- Normalization: Be cautious when comparing absolute abundances between DNA and RNA directly, as the number of ribosomes per cell is variable.
- 16S-Ratio (PSP): Calculate RNA:DNA ratios for individual taxa to estimate their Protein Synthesis Potential (PSP), a proxy for metabolic activity [113].
- Differential Abundance: Use tools like DESeq2 or LEfSe to identify taxa significantly enriched in the RNA fraction compared to the DNA fraction, indicating high activity.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key reagents and materials for implementing a combined DNA/RNA workflow
| Item | Function/Application | Example Product/Catalog Number |
|---|---|---|
| AllPrep DNA/RNA/miRNA Universal Kit | Simultaneous purification of genomic DNA and total RNA from a single sample. | Qiagen (Catalog number not specified in search results) |
| RLT Plus Buffer with DTT | Powerful lysis buffer that stabilizes RNA and DNA during sample storage and initial processing. | Qiagen [112] |
| DNase I, RNase-free | Degrades contaminating DNA in RNA samples prior to cDNA synthesis. | Various suppliers |
| SuperScript IV Reverse Transcriptase | High-sensitivity reverse transcription for cDNA synthesis from RNA templates. | Thermo Fisher Scientific |
| Pro341F / Pro805R Primers | Primer pair for amplification of the bacterial 16S V3-V4 hypervariable region. | Integrated DNA Technologies [112] |
| PNA Clamp | Peptide Nucleic Acid clamp to block amplification of host (e.g., mitochondrial) DNA in low-biomass samples. | PNA Bio Inc [112] |
| ZymoBIOMICS Microbial Community Standard | Defined mock community used as a positive control and for sensitivity validation. | Zymo Research (D6305) [112] |
Integrating RNA-based 16S rRNA transcript analysis with traditional DNA-based methods provides a powerful, multi-dimensional view of microbial communities. While DNA sequencing reveals the total taxonomic membership, RNA sequencing identifies the active subset of this community, often revealing critical drivers of ecosystem function that would otherwise remain hidden. The combined approach, supported by the detailed protocols and bioinformatics considerations outlined in this application note, enables researchers to move beyond mere census data and gain deeper insights into the dynamic and functionally active microbiome.
Within the context of a broader thesis on bioinformatics pipelines for 16S rRNA data analysis, the reproducibility of results across different computing environments emerges as a critical foundation for robust scientific discovery. Bioinformatics pipelines for microbial community analysis, such as QIIME and mothur, involve complex computational steps that could theoretically be influenced by the underlying operating system (OS). For researchers and drug development professionals, consistency in microbial taxonomic assignment and relative abundance estimates is paramount, as discrepancies could lead to divergent biological interpretations. This application note synthesizes empirical evidence evaluating the impact of operating systems on the reproducibility of 16S rRNA gene sequencing analysis, providing validated protocols and recommendations for ensuring cross-platform consistency in microbiome research.
Results
Quantitative Comparison of Pipeline Outputs Across Operating Systems
Table 1: Comparison of Bioinformatics Pipelines and Operating Systems on Taxonomic Abundance [86]
| Analysis Pipeline | Operating System | Bacteroides Relative Abundance (%) | Number of OTUs/ASVs | Inter-Platform Consistency |
|---|---|---|---|---|
| QIIME2 | Linux | 24.5 | Not Specified | Identical outputs between OS |
| QIIME2 | Mac | 24.5 | Not Specified | Identical outputs between OS |
| Bioconductor | Linux | 24.6 | Not Specified | Identical outputs between OS |
| Bioconductor | Mac | 24.6 | Not Specified | Identical outputs between OS |
| UPARSE | Linux | 23.6 | Not Specified | Minimal differences between OS |
| UPARSE | Mac | 20.6 | Not Specified | Minimal differences between OS |
| mothur | Linux | 22.2 | Not Specified | Minimal differences between OS |
| mothur | Mac | 21.6 | Not Specified | Minimal differences between OS |
A direct comparison of four bioinformatics pipelines run on both Linux and Mac operating systems revealed that pipeline choice has a more substantial impact on microbial community profiles than the operating system itself [86]. The study, analyzing 40 human stool samples, found that QIIME2 and Bioconductor provided identical outputs on both Linux and Mac OS, while UPARSE and mothur reported only minimal differences between operating systems [86]. Despite this cross-platform consistency, a statistically significant difference in relative abundance was observed for all phyla and the majority of abundant genera across pipelines, highlighting that studies using different pipelines cannot be directly compared without harmonization procedures [86].
Impact of Pipeline Selection on Microbial Community Analysis
Table 2: Performance Characteristics of Common 16S rRNA Analysis Pipelines [7] [17]
| Pipeline | Clustering Method | Sensitivity | Specificity | Richness Estimation | Best Application Context |
|---|---|---|---|---|---|
| QIIME-uclust | OTU (97%) | Low | Low | Inflated | Not recommended |
| MOTHUR | OTU (97%) | Moderate | Moderate | Higher for rare taxa | Full-control, custom analyses |
| USEARCH-UPARSE | OTU (97%) | Moderate | Moderate | Standard | Standardized OTU studies |
| DADA2 | ASV | High | Moderate | High resolution | Maximum sequence resolution |
| Qiime2-Deblur | ASV | Moderate | High | Standard | Balanced ASV approach |
| USEARCH-UNOISE3 | ASV | Moderate | High | Standard | Best balance for ASV studies |
Beyond operating system concerns, the fundamental choice of bioinformatics pipeline significantly influences analytical outcomes. A comprehensive evaluation of six bioinformatic pipelines on mock communities and a large fecal sample dataset (N=2,170) found that DADA2 offered the best sensitivity, while USEARCH-UNOISE3 showed the best balance between resolution and specificity [17]. The study recommended avoiding QIIME-uclust due to its production of spurious OTUs and inflation of alpha-diversity measures [17]. Another comparison focusing on rumen microbiota found that while both QIIME and mothur produced comparable results for abundant microorganisms, mothur assigned OTUs to a larger number of genera with higher relative abundance for less frequent microorganisms, particularly when using the GreenGenes database [7].
Experimental Protocols
Protocol: Validating Cross-Platform Consistency for Bioinformatics Pipelines
Experimental Design and Sample Preparation
- Sample Selection: Utilize a combination of mock microbial communities and actual experimental samples. The Human Microbiome Project Mock Community B (Even, Low concentration), containing 20 bacterial strains with known composition, is ideal for validation [17]. Include at least 40 biological samples to ensure statistical power, similar to the design used in prior validation studies [86].
- DNA Extraction and Sequencing: Perform standardized DNA extraction using kits such as the QIAamp DNA Stool Mini Kit with bead-beating homogenization [86]. Amplify the V3-V4 or V4 region of the 16S rRNA gene using Illumina-specific primers [86] [17]. Pool normalized libraries and sequence on an Illumina MiSeq platform with a minimum of 50,000 reads per sample after quality filtering [86] [7].
Bioinformatics Analysis Across Platforms
- Computing Environment Setup: Install identical versions of the target bioinformatics pipelines (QIIME2, mothur, UPARSE, DADA2) on Linux and Mac OS systems. For Linux, use a standardized distribution (e.g., Ubuntu 20.04 LTS). For Mac OS, use a consistent version (e.g., macOS Catalina or later) [86].
- Data Processing: Process the raw sequencing data from the same samples in parallel on both operating systems using the SILVA reference database (release 132) for all pipelines to minimize database-induced variation [86] [7]. Use default parameters for each pipeline unless specifically testing parameter sensitivity.
- Output Comparison: Generate operational taxonomic unit (OTU) or amplicon sequence variant (ASV) tables from each pipeline on each operating system. Compare the following outputs:
- Relative abundance of major phyla and genera (e.g., Bacteroides, Prevotella)
- Alpha diversity metrics (Shannon, Chao1, Observed OTUs/ASVs)
- Beta diversity measures (Bray-Curtis, Unweighted UniFrac distances)
- Taxonomic classification consistency at genus and species levels
Statistical Analysis and Validation
- Statistical Testing: Use the Friedman rank sum test to compare relative abundance differences across pipelines and operating systems, with post-hoc analysis for pairwise comparisons [86]. Assess cross-platform concordance using intraclass correlation coefficients for continuous measures and Cohen's kappa for categorical taxonomic assignments.
- Validation Criteria: Define acceptable limits of variation between operating systems (e.g., <1% difference in relative abundance for dominant taxa, >0.95 intraclass correlation for alpha diversity measures). Pipelines demonstrating >95% concordance in taxonomic assignments and non-significant differences (p > 0.05) in relative abundance between OS can be considered robust for cross-platform use [86].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for Cross-Platform Validation [86] [17] [7]
| Category | Item | Specification/Version | Function in Validation |
|---|---|---|---|
| Reference Materials | HM-782D Mock Community | HMP Mock Community B, Even | Provides known composition for validating pipeline accuracy and cross-platform consistency |
| Wet Lab Supplies | QIAamp DNA Stool Mini Kit | Standardized protocol | Ensures reproducible DNA extraction across samples intended for cross-platform comparison |
| Sequencing | Illumina MiSeq | V3 kit (2×300 bp) | Generates high-quality paired-end reads for 16S rRNA amplicon sequencing |
| Bioinformatics Pipelines | QIIME2 | 2019.10 or later | Modular, reproducible microbiome analysis with robust cross-platform performance |
| mothur | 1.39.5 or later | Comprehensive 16S analysis suite with minimal OS-dependent variations | |
| USEARCH/UPARSE | 11.0.667 or later | High-speed OTU clustering with good cross-platform consistency | |
| Reference Databases | SILVA | Release 132 or later | Provides high-quality aligned sequences for taxonomic classification, reducing database-induced bias |
| GreenGenes | 13_8 or later | Alternative reference database for taxonomic assignment |
Discussion
The empirical evidence demonstrates that modern bioinformatics pipelines exhibit generally good cross-platform consistency between Linux and Mac operating systems, with QIIME2 and Bioconductor showing identical outputs and other pipelines exhibiting only minimal differences [86]. This robustness across computing environments should provide confidence to research teams utilizing heterogeneous computational infrastructure. However, the choice of bioinformatics pipeline introduces significantly more variation in results than the operating system, necessitating careful pipeline selection based on study requirements [86] [17].
For research requiring maximum reproducibility, we recommend QIIME2 for its identical output across platforms and comprehensive reproducibility features [50]. When analyzing diverse communities with many rare taxa, mothur provides higher sensitivity for low-abundance organisms, though researchers should be aware of its slightly higher richness estimates [7]. For studies prioritizing exact sequence variants, USEARCH-UNOISE3 offers the best balance between resolution and specificity [17]. Critically, the same pipeline and reference database should be used throughout a study or across studies aiming for comparability, as harmonization between different pipelines remains challenging [86].
These findings underscore that while operating system choice is largely inconsequential for reproducible 16S rRNA analysis, standardization of the entire bioinformatics workflow is essential for generating comparable results across studies. Future methodology development should continue to prioritize computational reproducibility across diverse computing environments to advance microbiome science reliability.
The analysis of 16S rRNA gene sequencing data relies on bioinformatic pipelines to translate raw sequence data into biological insights. Tools like QIIME and mothur are central to this process, enabling the taxonomic classification of complex microbial communities [26]. However, the choice of computational methodology can significantly influence the resulting microbial composition, particularly affecting the detection and quantification of less abundant organisms [7] [8]. This application note synthesizes evidence on the performance of these pipelines, highlighting the consensus regarding core microbiota and the critical disagreements on low-abundance members. We further provide standardized protocols to enhance reproducibility and cross-study comparison in microbiome research, framed within the context of a broader thesis on 16S rRNA bioinformatics.
Synthesis of Comparative Findings
Consensus on Core Microbiota
Studies consistently demonstrate that different bioinformatics pipelines yield congruent results for the most abundant microbial taxa. Analyses of rumen and human fecal microbiota show that pipelines agree on the identity and relative abundance of dominant genera.
Table 1: Agreement on High-Abundance Genera in Rumen Microbiota (RA > 1%)
| Genus | QIIME Relative Abundance | mothur Relative Abundance | Statistical Significance (P-value) |
|---|---|---|---|
| Prevotella | High | High | > 0.05 |
| Succiniclasticum | High | High | > 0.05 |
| Butyrivibrio | High | High | > 0.05 |
| Methanobrevibacter | High | High | > 0.05 |
| Bifidobacterium | High | High | > 0.05 |
A separate study on human fecal samples confirmed this trend, finding that while the relative abundance estimates for phyla and the most abundant genera (e.g., Bacteroides) showed statistically significant differences between pipelines (P < 0.001), the overall taxonomic assignments were consistent [11]. This robust agreement on dominant community members provides confidence in cross-study comparisons of core microbiomes.
Disagreement on Less Abundant Members
In contrast to the stable core, the characterization of low-abundance microbiota is highly sensitive to the choice of bioinformatic pipeline and reference database. Significant differences emerge for taxa with a relative abundance below 10% [7] [8].
Table 2: Impact of Pipeline and Database on Low-Abundance Microbiota
| Parameter | QIIME with GreenGenes | mothur with GreenGenes | QIIME with SILVA | mothur with SILVA |
|---|---|---|---|---|
| Number of Genera (RA < 0.5%) | Lower | Higher | Intermediate | Intermediate |
| Analytical Sensitivity | Lower | Larger | Attenuated differences | Attenuated differences |
| Richness (Number of OTUs) | Lower (P < 0.05) | Larger (P < 0.05) | Comparable | Comparable |
| Impact on Beta Diversity | Significant | Significant | Reduced, but present | Reduced, but present |
When using the GreenGenes database, mothur consistently clustered a larger number of OTUs and assigned sequences to a greater number of genera in low abundance compared to QIIME [7] [8]. These differences directly distorted beta-diversity metrics, leading to different conclusions about the dissimilarity between microbial communities. The use of the SILVA database attenuated these discrepancies, though it did not completely eliminate them [7] [8] [11].
Experimental Protocols for Pipeline Comparison
To ensure reproducible and comparable microbiome analyses, the following detailed protocols are recommended.
Protocol 1: Benchmarking Pipeline Performance on a Mock Community
This protocol evaluates the accuracy and contamination sensitivity of a bioinformatics pipeline using a mock microbial community with a known composition.
Required Reagents and Materials:
- Mock Microbial Community: Composed of 8 bacterial species from 8 distinct genera (e.g., ATCC MSA-1002).
- DNA Dilution Buffers: Molecular grade water or TE buffer.
- Negative Control: Sterile, DNA-free water processed alongside mock samples.
Step-by-Step Procedure:
- Serial Dilution: Perform a serial 3-fold dilution of the mock community DNA across at least eight dilution points [115]. This creates a biomass gradient to simulate low-microbial-biomass conditions.
- Sequencing Library Preparation: Process all dilution points and the negative control through the standard 16S rRNA gene sequencing library preparation workflow (e.g., using Illumina's 16S Metagenomic Sequencing Library Preparation protocol) [115] [11].
- Bioinformatic Processing: Analyze the resulting sequencing data from all dilution points and the negative control using the bioinformatics pipeline(s) under evaluation (e.g., QIIME2, mothur).
- Computational Contamination Identification: Apply computational contaminant identification methods, such as the "frequency" method in the Decontam R package, which identifies sequences with an inverse correlation to DNA concentration [115].
- Performance Assessment:
- Calculate the percentage of contaminant sequences in each dilution (sequences not matching the mock community reference).
- Compare the observed microbial composition to the expected composition of the mock community to determine taxonomic accuracy.
- Evaluate alpha and beta diversity metrics across dilutions.
Protocol 2: Cross-Pipeline Analysis of Clinical Samples
This protocol standardizes the comparison of different bioinformatics pipelines using human-derived samples.
Required Reagents and Materials:
- Human Stool Samples: Collected in sterile containers and stored at -20°C until processing.
- DNA Extraction Kit: e.g., QIAamp DNA Stool Mini Kit with bead-beating homogenization [11].
- PCR Reagents: Primers targeting the V3-V4 hypervariable regions of the 16S rRNA gene [11].
Step-by-Step Procedure:
- DNA Extraction: Extract genomic DNA from ~200 mg of stool sample, including a mechanical lysis step (bead-beating for 10 min at 30 Hz) to ensure comprehensive cell disruption [11].
- 16S rRNA Gene Amplification & Sequencing: Amplify the target region (e.g., V3-V4) using standardized primers and cycling conditions. Purify amplicons and perform paired-end sequencing on an Illumina MiSeq platform [11].
- Multi-Pipeline Data Processing: Process the raw sequence data through each pipeline (e.g., QIIME2, mothur, UPARSE) to be compared.
- For all pipelines, use the same reference database (preferably SILVA) for taxonomic classification [7] [11].
- For QIIME2 and Bioconductor, use the DADA2 or Deblur plugin for Amplicon Sequence Variant (ASV) inference.
- For mothur and UPARSE, cluster sequences into Operational Taxonomic Units (OTUs) at 97% similarity.
- Data Harmonization and Comparison:
- Aggregate results at the genus level.
- Filter out genera with a relative abundance below 0.1% across all samples to focus on meaningful taxa [7].
- Compare the relative abundance of genera common to all pipelines using statistical tests (e.g., Friedman test). Assess the impact on downstream ecological metrics like alpha and beta diversity [11].
Visualizing the Experimental and Conceptual Workflow
Experimental Workflow for Pipeline Comparison
The following diagram illustrates the key steps for a robust cross-pipeline analysis, from sample preparation to data interpretation.
Conceptual Workflow of Pipeline Impact on Results
This diagram outlines the conceptual relationship between key methodological choices and their primary effects on the final microbiome analysis.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Materials for 16S rRNA Pipeline Research
| Item | Function/Benefit in Analysis |
|---|---|
| SILVA Reference Database | A curated, high-quality database often preferred over GreenGenes for classifying OTUs from diverse environments like the rumen, as it produces more comparable richness and diversity between pipelines [7] [11]. |
| Mock Microbial Community | A defined mix of known bacterial strains used as a positive control to evaluate pipeline accuracy, identify contaminants, and optimize bioinformatic parameters, especially in low-biomass contexts [115]. |
| Decontam R Package | A computational tool that uses sequence frequency or prevalence to identify and remove contaminant DNA sequences from 16S rRNA data, improving fidelity in low-biomass studies [115]. |
| QIAamp DNA Stool Mini Kit | A standardized DNA extraction kit that includes bead-beating for mechanical lysis, ensuring efficient and reproducible disruption of diverse bacterial cell walls in complex samples like stool [11]. |
| V3-V4 16S rRNA Primers | Primers targeting this specific hypervariable region are widely used (e.g., in Illumina's protocol) for bacterial community profiling, allowing for cross-study comparisons [11]. |
Conclusion
The choice between QIIME and mothur is not a matter of one being universally superior, but rather depends on the specific research question, sample type, and desired balance between sensitivity and consistency. While both pipelines reliably identify core, abundant microbial community members, significant differences arise in the estimation of relative abundances and the detection of low-abundance taxa, which are critically important in clinical settings. These discrepancies are often magnified by the choice of reference database, with SILVA frequently providing more consistent results. Future directions point towards the need for standardized harmonization procedures to enable direct cross-study comparisons and the integration of novel approaches like RNA-based sequencing to distinguish active from dormant community members. For biomedical research, a careful, validated, and well-documented bioinformatic strategy is as crucial as the wet-lab experiment itself to ensure that microbiome findings are robust, reproducible, and translatable into diagnostic and therapeutic applications.
References
- 1. 16S rRNA Sequencing Guide [https://blog.microbiomeinsights.com/16s-rrna-sequencing-guide]
- 2. 16S and ITS rRNA Sequencing | Identify bacteria & fungi ... [https://www.illumina.com/areas-of-interest/microbiology/microbial-sequencing-methods/16s-rrna-sequencing.html]
- 3. Research progress on the application of 16S rRNA gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10869621/]
- 4. Nanopore full length 16S rRNA gene sequencing ... [https://www.nature.com/articles/s41598-025-10999-8]
- 5. Full-length 16S rRNA gene sequencing by PacBio improves ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10213-5]
- 6. mothur and QIIME [https://mothur.org/blog/2016/mothur-and-qiime/]
- 7. Comparison of Mothur and QIIME for the Analysis ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2018.03010/full]
- 8. Comparison of Mothur and QIIME for the Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6300507/]
- 9. Next Generation Sequencing Improves Diagnostic 16S ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12418051/]
- 10. Transformative biomedical research program exposes ... [https://www.unr.edu/nevada-today/news/2025/microbiome-undergraduate-summer-bootcamp]
- 11. Comparison of Bioinformatics Pipelines and Operating ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.01262/full]
- 12. QIIME 2 2025.4 is now available! [https://qiime2.org/news/qiime-2-2025-4-is-now-available-33088]
- 13. A detailed workflow to develop QIIME2-formatted reference ... [https://bmcgenomdata.biomedcentral.com/articles/10.1186/s12863-022-01067-5]
- 14. [Preview] QIIME 2 2025.10 development changelog [https://forum.qiime2.org/t/preview-qiime-2-2025-10-development-changelog/33589]
- 15. Comparison of Mothur and QIIME for the Analysis ... [https://pubmed.ncbi.nlm.nih.gov/30619117/]
- 16. The unresolved struggle of 16S rRNA amplicon sequencing [https://environmentalmicrobiome.biomedcentral.com/articles/10.1186/s40793-025-00705-6]
- 17. Comparing bioinformatic pipelines for microbial 16S rRNA ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0227434]
- 18. QIIME2 enhances multi-amplicon sequencing data analysis [https://pubmed.ncbi.nlm.nih.gov/40711419/]
- 19. Microbiome Informatics: OTU vs. ASV [https://www.zymoresearch.com/blogs/blog/microbiome-informatics-otu-vs-asv?srsltid=AfmBOopG2Zn-qNcu-_nfBuEfOXi8163chNSXq5I9GS19JIR7whe3ilCp]
- 20. OTUs and ASVs Produce Comparable Taxonomic and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8070570/]
- 21. 16s data processing and microbiome analysis - GitHub Pages [https://h3abionet.github.io/H3ABionet-SOPs/16s-rRNA]
- 22. Introduction to Amplicon Sequence Variants [https://www.cd-genomics.com/resource-introduction-amplicon-sequence-variants.html]
- 23. Considerations and best practices in animal science 16S ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8807179/]
- 24. 16S rRNA Gene Sequencing vs. Shotgun Metagenomic ... [https://blog.microbiomeinsights.com/16s-rrna-sequencing-vs-shotgun-metagenomic-sequencing]
- 25. 16S rRNA Sequencing VS Shotgun ... [https://www.cd-genomics.com/microbioseq/resource-16s-rrna-sequencing-vs-shotgun-sequencing-for-microbial-research]
- 26. The Human Microbiome and Understanding the 16S rRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5535273/]
- 27. 16S rRNA WORKFLOW REPORT - MBBU [https://mbbu.github.io/16S-mini-project/]
- 28. Galaxy mothur Toolset (GmT): a user-friendly application for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6377400/]
- 29. Comparison between 16S rRNA and shotgun sequencing in ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10621-7]
- 30. Amplicon-Based Next-Generation Sequencing vs. ... [https://www.cd-genomics.com/microbioseq/amplicon-based-next-generation-sequencing-vs-metagenomic-shotgun-sequencing.html]
- 31. Comparison between 16S rRNA and shotgun sequencing ... [https://www.nature.com/articles/s41598-021-82726-y]
- 32. Analysis of the microbiome: Advantages of whole genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4830092/]
- 33. Evaluation of 16S rRNA gene sequencing for species and ... [https://www.nature.com/articles/s41467-019-13036-1]
- 34. , Pipelines, Parameters: Issues in Primer Gene 16 ... S rRNA Sequencing [https://pmc.ncbi.nlm.nih.gov/articles/PMC8544895/]
- 35. Primer and platform effects on 16S rRNA tag sequencing [https://pmc.ncbi.nlm.nih.gov/articles/PMC4523815/]
- 36. Development of an Analysis Pipeline Characterizing... | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0148047]
- 37. Unveiling the impact of 16S rRNA gene intergenomic ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1573920/full]
- 38. Optimizing PCR primers targeting the bacterial 16S ribosomal ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2360-6]
- 39. 16S rRNA gene primer choice impacts off-target ... [https://www.nature.com/articles/s41598-023-39575-8]
- 40. The Effect of Primer Choice and Short Read Sequences on ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0071360]
- 41. Non-specific amplification of human DNA is a major ... [https://www.nature.com/articles/s41598-020-73403-7]
- 42. Direct 16 -seq from bacterial communities: a PCR-independent... S rRNA [https://www.nature.com/articles/srep32165?error=cookies_not_supported&code=190d6ed4-c978-4770-a32a-ffa5a23f68e1]
- 43. MiSeq SOP [https://mothur.org/wiki/miseq_sop/]
- 44. Development of a Dual-Index Sequencing Strategy and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3753973/]
- 45. 16S Microbial Analysis with mothur (extended) [https://training.galaxyproject.org/training-material/topics/microbiome/tutorials/mothur-miseq-sop/tutorial.html]
- 46. 16S Pipeline in Mothur - Ariana S. Huffmyer, PhD [https://ahuffmyer.github.io/ASH_Putnam_Lab_Notebook/16S-Analysis-in-Mothr-Part-1/]
- 47. Inquiry on 16S rRNA Analysis Using Mothur and Data ... [https://forum.mothur.org/t/inquiry-on-16s-rrna-analysis-using-mothur-and-data-trimming-techniques/22333]
- 48. Qiime2 tutorial for Cris - Gabriel M. [https://guto-monteiro.com/blog/2025/blog-post-9/]
- 49. Overview of QIIME 2 Plugin Workflows [https://docs.qiime2.org/2024.10/tutorials/overview/]
- 50. Comprehensive end-to-end microbiome analysis ... - qiime 2 [https://curr-protoc-bioinformatics.qiime2.org/]
- 51. Conceptual overview of marker gene (i.e., amplicon) data ... [https://amplicon-docs.qiime2.org/en/2025.4/explanations/conceptual-overview.html]
- 52. QIIME 2 for experienced microbiome researchers [https://amplicon-docs.qiime2.org/en/latest/explanations/experienced-researchers.html]
- 53. dada2 - Microbiome marker gene analysis with QIIME 2 [https://amplicon-docs.qiime2.org/en/2025.7/references/plugins/dada2.html]
- 54. Bioinformatics Beginner Needs help - User Support [https://forum.qiime2.org/t/bioinformatics-beginner-needs-help/4226]
- 55. “Moving Pictures” tutorial - Multiple Interface Edition [https://docs.qiime2.org/2024.10/tutorials/moving-pictures-usage/]
- 56. QIIME 2 Enables Comprehensive End‐to‐End Analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9285460/]
- 57. Reconciling 16s Databases in a Single Pipeline [https://forum.qiime2.org/t/reconciling-16s-databases-in-a-single-pipeline/31258]
- 58. 16S rRNA Methodology and Analysis Guide [https://cores.emory.edu/eicc/_includes/documents/sections/resources/16s_methodology_2024-01-24.html]
- 59. Reprocessing 16S rRNA Gene Amplicon Sequencing ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2021.720637/full]
- 60. Pre-processing reads - 16S rRNA analysis [https://rachaellappan.github.io/16S-analysis/pre-processing-reads.html]
- 61. PEAR - Paired-end read merger [https://cme.h-its.org/exelixis/web/software/pear/doc.html]
- 62. Chimeric 16S rRNA sequence formation and detection in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3044863/]
- 63. The Effects of Alignment Quality, Distance Calculation Method ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1000844]
- 64. Filtering 16S rRNA sequencing data for quality [https://bio-protocol.org/exchange/minidetail?id=3415860&type=30]
- 65. OTU vs. ASV in Metagenomic Analysis [https://www.seqme.eu/en/blog/article/otu-vs-asv-in-metagenomic-analysis]
- 66. The choice of OTUs vs. ASVs in 16S rRNA amplicon data ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0264443]
- 67. Comparing bioinformatic pipelines for microbial 16S rRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6964864/]
- 68. Ranking the biases: The choice of OTUs vs. ASVs in 16S ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8870492/]
- 69. Microbiome Informatics: OTU vs. ASV [https://www.zymoresearch.com/blogs/blog/microbiome-informatics-otu-vs-asv?srsltid=AfmBOoqBQ4yUq7WOuBTYopU7CbpuYyJM6SVx3YqgHAziUh-ObFVElPlc]
- 70. Evaluation of 16S rRNA Databases for Taxonomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6440677/]
- 71. SILVA: Silva [https://www.arb-silva.de/]
- 72. The SILVA ribosomal RNA gene database project [https://pmc.ncbi.nlm.nih.gov/articles/PMC3531112/]
- 73. SILVA taxonomy [https://www.arb-silva.de/documentation/silva-taxonomy]
- 74. SILVA, RDP, Greengenes, NCBI and OTT — how do these ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-3501-4]
- 75. Greengenes, a Chimera-Checked 16S rRNA Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1489311/]
- 76. Greengenes-formatted databases [https://mothur.org/wiki/greengenes-formatted_databases/]
- 77. Introducing Greengenes2 2022.10 [https://forum.qiime2.org/t/introducing-greengenes2-2022-10/25291]
- 78. Beta diversity - 16S rRNA analysis - Rachael Lappan [https://rachaellappan.github.io/16S-analysis/beta-diversity.html]
- 79. QIIME Overview Tutorial — Homepage [http://qiime.org/1.3.0/tutorials/tutorial.html]
- 80. Moving Pictures tutorial 🎥 - Microbiome marker gene ... [https://amplicon-docs.qiime2.org/en/latest/tutorials/moving-pictures.html]
- 81. Alpha and Beta Diversity Explanations and Commands [https://forum.qiime2.org/t/alpha-and-beta-diversity-explanations-and-commands/2282]
- 82. how to test significant difference between diversity indexes? [https://forum.mothur.org/t/how-to-test-significant-difference-between-diversity-indexes/3223]
- 83. 16S rRNA sequencing analysis: the devil is in the details [https://pmc.ncbi.nlm.nih.gov/articles/PMC7524321/]
- 84. Updating the 97% Identity Threshold for 16S Ribosomal ... [https://pubmed.ncbi.nlm.nih.gov/29506021/]
- 85. Using QIIME to Analyze 16S rRNA Gene Sequences from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4477843/]
- 86. Comparison of Bioinformatics Pipelines and Operating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7318847/]
- 87. The EMP, QIIME and Mothur [https://forum.mothur.org/t/the-emp-qiime-and-mothur/1425]
- 88. Primer selection impacts specific population abundances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6175402/]
- 89. CONSTAX: a tool for improved taxonomic resolution of ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1952-x]
- 90. 16S rRNA metabarcoding applied to the microbiome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11904769/]
- 91. Research Note: Choice of microbiota database affects data ... [https://www.sciencedirect.com/science/article/pii/S0032579122002632]
- 92. GSR-DB: a manually curated and optimized taxonomical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10946287/]
- 93. Comparing QIIME2 and Mothur 16S QC and Filtering [https://ahuffmyer.github.io/ASH_Putnam_Lab_Notebook/Comparing-QIIME2-and-Mothur-16S-QC-and-Filtering/]
- 94. Greengenes2 unifies microbial data in a single reference tree [https://www.nature.com/articles/s41587-023-01845-1]
- 95. Guidelines for preventing and reporting contamination in ... [https://www.nature.com/articles/s41564-025-02035-2]
- 96. an R package for decontaminating low-biomass 16S-rRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12095030/]
- 97. Quantitative profiling of bacterial communities via full length ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-025-04399-1]
- 98. Ultrafast and accurate 16S rRNA microbial ... - Microbiome [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-020-00900-2]
- 99. The unresolved struggle of 16S rRNA amplicon sequencing [https://pmc.ncbi.nlm.nih.gov/articles/PMC12076876/]
- 100. Using short-read 16S rRNA sequencing of multiple ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11118338/]
- 101. Longitudinal profiling of low-abundance strains in ... [https://www.nature.com/articles/s41564-025-01983-z]
- 102. Extending and improving metagenomic taxonomic profiling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10635831/]
- 103. Bioinformatics Analysis of 16S rRNA Amplicon Sequencing [https://www.cd-genomics.com/bioinformatics-analysis-of-16s-rrna-amplicon-sequencing.html]
- 104. A framework for assessing 16S rRNA marker-gene survey ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7071580/]
- 105. Using QIIME to analyze 16S rRNA gene sequences from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3249058/]
- 106. Using Mothur to Determine Bacterial ... [https://pubmed.ncbi.nlm.nih.gov/31524992/]
- 107. Week 8: Classifying taxonomy of short reads with mothur V.1 [https://www.protocols.io/view/week-8-classifying-taxonomy-of-short-reads-with-mo-g7tbznn.html]
- 108. How to choose between full-length Greengenes or SILVA ... [https://forum.qiime2.org/t/how-to-choose-between-full-length-greengenes-or-silva-for-16s-rrna-v1-v2-analysis-with-qiime2-pipeline/31240]
- 109. What is the difference between Greengenes and SILVA? [https://forum.qiime2.org/t/what-is-the-difference-between-greengenes-and-silva/1560]
- 110. 16S rRNA gene sequencing of mock microbial populations [https://pmc.ncbi.nlm.nih.gov/articles/PMC4921037/]
- 111. Possible Analysis Pipeline for Ion Torrent 16S ... [https://forum.qiime2.org/t/possible-analysis-pipeline-for-ion-torrent-16s-metagenomics-kit-data-in-qiime2/13476]
- 112. Comparison of RNA- and DNA-based 16S amplicon ... [https://pubmed.ncbi.nlm.nih.gov/40379732/]
- 113. A comparison of 16S rRNA-gene and 16S rRNA-transcript ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1608399/full]
- 114. Comparison of RNA- and DNA-based 16S amplicon ... [https://www.nature.com/articles/s41598-025-00969-5]
- 115. Controlling for Contaminants in Low-Biomass 16S rRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6550369/]