Optimized Flow Cytometry Protocol for Intracellular Stem Cell Markers: A Guide from Foundational Principles to Advanced Validation
This article provides a comprehensive guide for researchers and drug development professionals on flow cytometry protocols for analyzing intracellular stem cell markers.
Optimized Flow Cytometry Protocol for Intracellular Stem Cell Markers: A Guide from Foundational Principles to Advanced Validation
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on flow cytometry protocols for analyzing intracellular stem cell markers. It covers foundational principles of stem cell marker biology and intracellular staining, detailed step-by-step methodologies for sample preparation and staining, essential troubleshooting for common issues like high background and weak signals, and rigorous validation techniques to ensure reproducible and accurate data. By integrating strategic planning with practical optimization tips, this resource supports the reliable characterization of pluripotent stem cells, crucial for advancements in regenerative medicine and disease modeling.
Understanding Stem Cell Marker Biology and Intracellular Staining Fundamentals
Pluripotency is the defining characteristic of a cell that possesses the capability to self-renew indefinitely and differentiate into any cell type derived from the three primary germ layers. This fundamental process is indispensable during organogenesis in fetal development and throughout tissue repair in health and disease. The pluripotent state is rigorously controlled by a core group of transcription factors (TFs) that form an intricate regulatory network. In this network, NANOG, OCT4 (POU5F1), and SOX2 function as master regulators, governing the transcriptional programs that maintain self-renewal and suppress differentiation. Their discovery, particularly that of NANOG, has profoundly advanced our understanding of stem cell biology. The gene was aptly named after Tír Na nÓg, the "Land of Eternal Youth" from Irish mythology, reflecting its crucial role in maintaining a youthful, undifferentiated state in cells [1].
These core TFs function not in isolation but through a collaborative mechanism, binding to thousands of genes to activate those necessary for pluripotency and repress those involved in differentiation. This article details the critical roles of these intracellular markers, with a specific focus on NANOG, and provides detailed methodologies for their detection using flow cytometry, a powerful technique for single-cell analysis. This content is framed within a broader thesis on flow cytometry protocols for intracellular stem cell marker research, providing researchers and drug development professionals with both theoretical knowledge and practical application guidelines.
Core Pluripotency Transcription Factors
The core pluripotency transcription factors, OCT4, SOX2, and NANOG, constitute the central regulatory circuitry that maintains embryonic stem cell (ESC) identity. They achieve this through a complex, interconnected autoregulatory loop and by co-occupying the promoters of a vast array of target genes.
NANOG: The Master Sustainer of Pluripotency
NANOG is a divergent homeodomain protein that serves as a pivotal sustainer of pluripotency.
- Gene and Protein Structure: The human NANOG gene is located on chromosome 12p13.31. It encodes a 305-amino acid protein with a molecular weight of approximately 34.6 kDa. Its structure includes a well-conserved Nk-2 homeodomain (amino acids 95-155) responsible for DNA binding, and a unique C-terminal domain featuring a tryptophan repeat (WR) that is critical for protein dimerization and interaction with other pluripotency network proteins [1].
- Functional Role: NANog's primary function is to sustain the pluripotent state by resisting or reversing differentiation signals. It maintains the pluripotency of ESCs even in the absence of the Leukemia Inhibitory Factor (LIF)/STAT3 pathway, a key signaling axis for self-renewal. While OCT4 levels must be tightly regulated—as both upregulation and downregulation induce differentiation—elevated NANOG expression promotes LIF-independent self-renewal and inhibits differentiation into extra-embryonic endoderm [1] [2]. This positions NANOG as a "master" specifier of ES cell identity. Notably, while NANOG is crucial, some studies indicate that embryonic stem cells can self-renew indefinitely in its permanent absence, though they become predisposed to differentiate. Its role appears most critical in the inner cell mass and, indispensably, in germ cell development [3].
- Regulation and Signaling: The expression of NANOG itself is regulated by OCT4 and SOX2, which bind to its promoter region. NANOG, in turn, helps maintain the pluripotency network by blocking differentiation signals, such as Bone Morphogenetic Protein (BMP)-induced differentiation, through its interaction with Smad1 [4]. It also operates through signaling pathways like JAK/STAT and Wnt/β-catenin to promote stemness [5].
OCT4 and SOX2: Essential Collaborators
OCT4 and SOX2 are the other two pillars of the core pluripotency network.
- OCT4 (POU5F1): A POU-family transcription factor essential for the formation of the inner cell mass (ICM) and the maintenance of ESC pluripotency. Its expression levels are exquisitely sensitive; a >50% reduction causes differentiation into trophectoderm, while a >50% increase drives differentiation into primitive endoderm and mesoderm [2] [6].
- SOX2: An HMG-box transcription factor that frequently partners with OCT4. Together, they bind to composite SOX/OCT elements in the genome to regulate a wide array of target genes, including NANOG. SOX2 also plays a critical "bookmarking" role during mitosis, helping to re-establish the pluripotent gene expression program after cell division [6].
The table below summarizes the key characteristics of these core pluripotency transcription factors.
Table 1: Core Pluripotency Transcription Factors
| Transcription Factor | Gene Family | Key Functions in Pluripotency | Consequence of Downregulation |
|---|---|---|---|
| NANOG | Divergent Homeodomain | Sustains LIF-independent self-renewal; blocks BMP-driven differentiation; "naïve" pluripotency TF [1] [6] [4] | Differentiation into extra-embryonic endoderm lineages [2] |
| OCT4 (POU5F1) | POU-domain | Regulates cell fate in early embryo; essential for ICM and ESC identity [1] [2] | Differentiation into trophectoderm [2] |
| SOX2 | HMG-box | Partners with OCT4; mitotic bookmarker; maintains epiblast [1] [6] | Loss of pluripotency; impaired self-renewal [2] |
The Pluripotency Network and Dynamics
The core TFs do not operate in a static environment. Their expression and nuclear organization are highly dynamic and influenced by the cell cycle and external cues.
- Transcriptional Network: Genome-wide studies have shown that OCT4, SOX2, and NANOG co-occupy the promoters of a substantial number of their own genes and many other developmental regulators. This creates a robust, interconnected network that reinforces the pluripotent state while suppressing the expression of genes associated with differentiation [4].
- Spatial Organization and Cell Cycle: Recent research reveals that these TFs partition into biomolecular condensates or foci within the nucleus of ESCs. The organization of these condensates changes during the cell cycle, particularly during S-phase when DNA replication occurs. Furthermore, differentiation cues received during the G1 phase trigger a rapid and massive reorganization of these condensates in the subsequent early-S phase, with OCT4 and SOX2 foci dissolving and their chromatin interaction dynamics altering, preceding their downregulation [6]. NANOG, as a naïve pluripotency marker, shows impaired chromatin interactions upon early differentiation stimuli [6].
The following diagram illustrates the core regulatory circuitry and the key external signals that support it.
Diagram 1: The Core Pluripotency Network. External signals (LIF, BMP, WNT) activate intracellular pathways that converge on the core transcription factors OCT4, SOX2, and NANOG. These TFs form an interconnected auto-regulatory loop and collectively regulate genes responsible for self-renewal and pluripotency. Solid lines indicate direct activation; dashed lines represent indirect or context-dependent interactions [1] [4].
Flow Cytometry for Intracellular Transcription Factor Analysis
Flow cytometry is an indispensable tool for quantifying and characterizing intracellular stem cell markers at the single-cell level. Unlike cell surface staining, intracellular staining for transcription factors requires specific protocols to maintain cell integrity while allowing antibodies access to nuclear targets.
Key Principles of Intracellular Staining
The successful detection of intracellular proteins like NANOG, OCT4, and SOX2 hinges on two critical steps: fixation and permeabilization.
- Fixation: This process uses cross-linking agents (e.g., formaldehyde) to preserve the cell's structural integrity and immobilize intracellular antigens, preventing their degradation or loss during subsequent handling.
- Permeabilization: This step creates pores in the cellular and nuclear membranes using detergents or alcohol-based buffers, allowing fluorescently-conjugated antibodies to enter the cell and bind to their specific intracellular targets [7] [8].
The optimal fixation and permeabilization method depends heavily on the subcellular location and nature of the target protein. The table below compares the common buffer systems used for different intracellular targets.
Table 2: Fixation and Permeabilization Buffer Systems for Intracellular Targets
| Buffer System Type | Primary Use | Examples (Commercial Kits) | Key Considerations |
|---|---|---|---|
| Mild Detergent-Based | Cytoplasmic proteins, cytokines, secreted proteins [8] | BD Cytofix/Cytoperm Buffer Set; FIX & PERM Kit [7] [8] | Not recommended for intranuclear proteins. Can preserve some cell surface markers. |
| Harsh Alcohol-Based | Phosphorylated proteins (phospho-epitopes) [7] | BD Phosflow Perm Buffer III [7] | Can denature many cell surface antigens and some intracellular proteins. |
| Transcription Factor-Specific | Nuclear transcription factors (e.g., NANOG, OCT4, SOX2, FoxP3) [7] [8] | BD Pharmingen Transcription Factor Buffer Set; eBioscience Foxp3/Transcription Factor Staining Buffer Set [7] [8] | Formulated to disrupt nuclear complexes and allow antibody access to DNA-bound TFs. Compatibility with surface markers varies. |
Detailed Protocol: Staining for NANOG and Other Pluripotency TFs
The following is a detailed protocol for the simultaneous detection of cell surface markers and intracellular transcription factors like NANOG in suspended cells, adapted from manufacturer guidelines [7] [9] [8].
Pre-Staining: Sample Preparation
- Harvest Cells: Harvest and wash cells (e.g., ESCs, differentiated progeny) in a cold PBS buffer supplemented with 0.5%–1% BSA.
- Count and Aliquot: Count the cells and aliquot up to 1–2 x 10^6 cells per flow cytometry tube. Centrifuge and decant the supernatant.
- Viability Staining (Optional but Recommended): Resuspend the cell pellet in a fixable viability dye (e.g., LIVE/DEAD Fixable Stain) diluted in PBS. Incubate for 15-30 minutes on ice in the dark. Wash cells with 2 mL of flow cytometry staining buffer to remove excess dye [8].
- Fc Receptor Blocking: To reduce non-specific antibody binding, resuspend the cell pellet in an Fc receptor blocking solution (e.g., purified IgG) and incubate for 10-15 minutes at room temperature [9].
Surface Staining
- Add Surface Antibodies: Directly add fluorochrome-conjugated antibodies against cell surface markers (e.g., SSEA-4, TRA-1-60 for human pluripotent stem cells) to the tube. Vortex gently and incubate for 30 minutes at room temperature in the dark.
- Wash: Add 2 mL of flow cytometry staining buffer, centrifuge, and decant the supernatant. Repeat this wash step twice to ensure the removal of unbound antibodies [9].
Intracellular Staining for Transcription Factors
- Fix and Permeabilize: Thoroughly resuspend the cell pellet in a commercial transcription factor fixation/permeabilization buffer (e.g., from the BD Pharmingen Transcription Factor Buffer Set). Incubate for 30-60 minutes at room temperature in the dark.
- Wash with Permeabilization Buffer: Add 2 mL of a 1X permeabilization wash buffer. Centrifuge, decant the supernatant. Note: The supernatant may contain fixative, so dispose of it according to safety regulations.
- Intracellular Antibody Staining: Resuspend the fixed and permeabilized cells in permeabilization buffer containing pre-titrated, fluorochrome-conjugated antibodies against intracellular targets (e.g., anti-NANOG, anti-OCT4, anti-SOX2). Incubate for 30-60 minutes at room temperature in the dark.
- Final Washes: Wash the cells twice with 2 mL of permeabilization buffer to remove unbound intracellular antibodies.
- Resuspension and Analysis: Resuspend the final cell pellet in 200–400 µL of flow cytometry staining buffer for acquisition on the flow cytometer.
The workflow for this protocol is summarized in the following diagram.
Diagram 2: Workflow for Staining Intracellular Transcription Factors. The protocol involves sequential steps: cell preparation, surface marker staining, fixation/permeabilization, and finally, intracellular staining for nuclear transcription factors like NANOG [7] [9] [8].
The Scientist's Toolkit: Essential Reagents and Materials
The following table lists key reagents and materials required for the successful intracellular staining of pluripotency transcription factors.
Table 3: Essential Research Reagents for Intracellular Flow Cytometry
| Item Category | Specific Examples | Function |
|---|---|---|
| Fixation/Permeabilization Kits | BD Pharmingen Transcription Factor Buffer Set (Cat. No. 562574/562725); eBioscience Foxp3/Transcription Factor Staining Buffer Set (Cat. No. 00-5523-00) [7] [8] | To simultaneously fix cells and permeabilize nuclear membranes for antibody access to TFs. |
| Flow Cytometry Staining Buffer | Flow Cytometry Staining Buffer (e.g., R&D Systems Catalog # FC001); PBS with 0.5%–5% BSA or FBS [9] | To wash cells and dilute antibodies while maintaining cell viability and reducing non-specific binding. |
| Viability Dyes | LIVE/DEAD Fixable Dead Cell Stains (Various fluorochromes) [8] | To distinguish and exclude dead cells from analysis, improving data accuracy. |
| Fc Receptor Blocking Reagent | Purified anti-CD16/32 (for mouse cells); Human Fc Receptor Binding Inhibitor; Purified IgG [9] | To block Fc receptors on cells, minimizing non-specific antibody binding. |
| Antibodies | Fluorochrome-conjugated antibodies against NANOG, OCT4, SOX2, and relevant cell surface markers. | To specifically detect and label target antigens for fluorescence detection. |
| Compensation Controls | UltraComp eBeads [8] | To create single-color controls for accurate fluorescence compensation. |
| Cell Lines & Controls | Validated pluripotent stem cells (e.g., mouse ESCs W4, YPet-OCT4 ESCs) [6] | To provide positive and negative biological controls for staining optimization. |
Application in Stem Cell Biology and Disease Research
The analysis of intracellular pluripotency markers extends beyond basic biology into critical applications in disease modeling and drug development.
- Monitoring Pluripotency in Real-Time: The dynamic organization of OCT4, SOX2, and NANOG condensates, and their changes during the cell cycle, provides insights into the stability of the pluripotent state. Differentiation cues trigger a rapid dissolution of these condensates in early-S phase, an event that can be tracked using advanced fluorescence techniques [6]. This makes them sensitive indicators of the initial steps toward cell fate commitment.
- Quality Control in Stem Cell Culture and Differentiation: Flow cytometry for NANOG, OCT4, and SOX2 is a gold-standard method for quantifying the proportion of undifferentiated cells in a culture. It is also used to optimize and assess the efficiency of differentiation protocols by tracking the downregulation of these pluripotency markers alongside the upregulation of lineage-specific markers [7].
- Cancer Stem Cell (CSC) Research: NANOG is frequently found at high levels in various cancers (e.g., leukemia, liver, colorectal, prostate, ovarian, lung, and breast cancers) [5]. Its upregulation is strongly associated with advanced disease stages and poor prognosis. In CSCs, NANog promotes stemness, self-renewal, metastasis, invasiveness, and chemoresistance through pathways like JAK/STAT and Wnt/β-catenin [5]. Therefore, targeting NANOG and the CSC population is a promising therapeutic strategy.
The core pluripotency transcription factors NANOG, OCT4, and SOX2 form the bedrock of our understanding of stem cell identity and fate. Their intricate network, dynamic behavior, and pivotal functions underscore their importance as key intracellular markers. The detailed flow cytometry protocols provided here, including specific reagent recommendations and a step-by-step workflow, offer researchers a robust methodological framework for investigating these markers. The ability to accurately detect and quantify these proteins is fundamental to advancing research in regenerative medicine, understanding the mechanisms of pluripotency, and developing novel therapies that target stem cells in diseases like cancer.
The Critical Importance of Single-Cell Suspensions for Accurate Flow Cytometry Analysis
Flow cytometry has established itself as a versatile and powerful tool in stem cell research, enabling the high-throughput, multi-parameter analysis essential for identifying and characterizing rare stem cell populations within heterogeneous samples [10]. The technique's capability to rapidly analyze thousands of cells per second and isolate even rare stem cells through fluorescence-activated cell sorting (FACS) hinges on a fundamental prerequisite: the sample must be a high-quality single-cell suspension [10] [11]. Clumps or aggregated cells can obstruct the fluidics system of the cytometer, cause erratic fluid stream behavior, and lead to inaccurate data interpretation by registering multiple cells as a single event. For intracellular staining of stem cell markers—a common requirement for assessing pluripotency—the need for optimal single-cell suspensions becomes even more critical, as fixation and permeabilization steps can exacerbate cell clumping [11] [12]. This application note details the protocols and quantitative assessments necessary to prepare superior single-cell suspensions, ensuring reliable and reproducible flow cytometric data for intracellular stem cell marker research.
Quantitative Assessment of Single-Cell Suspension Quality
The quality of a single-cell suspension directly impacts all downstream analyses. The following parameters should be assessed and optimized prior to flow cytometry.
Table 1: Key Parameters for Assessing Single-Cell Suspension Quality
| Parameter | Target Value | Measurement Technique | Impact on Flow Cytometry Data |
|---|---|---|---|
| Cell Viability | >90-95% [11] | Viability dye (e.g., 7-AAD, DAPI) [11] | Reduces non-specific antibody binding and false-positive events [11]. |
| Clump Frequency | Minimized | Microscopic examination, light scatter plot analysis [11] | Prevents instrument clogging and misidentification of cell doublets as single events. |
| Cell Concentration | 0.5–1 x 10⁶ cells/mL [11] | Hemocytometer or automated cell counter | Ensures event rate is within instrument's optimal acquisition range. |
| Post-Fixation Clumping | Minimized | Light scatter plot analysis post-fixation [12] | Maintains single-cell status after chemical treatment, crucial for intracellular staining. |
Comprehensive Protocols for Sample Preparation
Generation of Single-Cell Suspensions from Cell Cultures
This protocol is designed for adherent stem cell cultures, such as induced pluripotent stem cells (iPSCs).
Basic Protocol 1: iPSC Culture and Collection for Flow Cytometry Analysis [13]
- Culture and Harvest: Grow iPSCs to approximately 70-80% confluence.
- Enzyme Dissociation: Aspirate the culture medium and wash cells gently with phosphate-buffered saline (PBS). Add an appropriate volume of cell dissociation reagent (e.g., Accutase or Trypsin-EDTA) and incubate at 37°C until cells detach.
- Neutralization: Neutralize the dissociation enzyme with complete medium containing serum.
- Wash and Filter: Centrifuge the cell suspension at ~200 x g for 5 minutes at 4°C. Resuspend the cell pellet in an ice-cold suspension buffer (e.g., PBS with 5-10% fetal calf serum). Pass the suspension through a 40-70 µm cell strainer to remove any remaining clumps.
- Count and Assess Viability: Determine cell concentration and viability using a hemocytometer with Trypan Blue exclusion or an automated cell counter. Adjust concentration to 0.5–1 x 10⁶ cells/mL for staining.
Concomitant Staining of Surface and Intracellular Markers
For stem cell characterization, it is often necessary to co-stain surface markers (e.g., CD34 for hematopoietic stem cells) and intracellular markers (e.g., transcription factors like NANOG) [10] [13]. The following workflow and protocol outline a simultaneous staining method that minimizes cell loss.
Basic Protocol 2: Staining of iPSCs for Extracellular and Intracellular Undifferentiated Stem Cell Markers [13]
Materials:
- Suspension/Wash Buffer: PBS with 5-10% fetal calf serum (FCS)
- Fixative: 4% Paraformaldehyde (PFA), ice-cold [11] [12]
- Permeabilization Solution: 90% Methanol (ice-cold) or 0.1% Triton X-100 in PBS [11] [12]
- Antibodies: Fluorochrome-conjugated antibodies against surface and intracellular targets.
- FcR Blocking Buffer: e.g., 2-10% goat serum or human IgG [11]
Steps:
- Live/Dead Staining (Optional but Recommended): Resuspend the single-cell suspension in a viability dye according to the manufacturer's protocol. Wash cells twice with wash buffer [11].
- Fc Receptor Blocking: Resuspend the cell pellet in an appropriate blocking buffer and incubate for 30-60 minutes in the dark at 4°C to prevent non-specific antibody binding. Wash cells twice [11].
- Cell Surface Staining (Optional): For surface markers that are sensitive to fixation/permeabilization, stain live cells with the conjugated antibody at this stage. Wash cells twice. Note: If using methanol for permeabilization, avoid protein-based fluorophores (e.g., PE, APC) here as they will be denatured [12].
- Fixation: Resuspend the cell pellet in 100 µL of ice-cold 4% PFA. Gently vortex and incubate for 15-20 minutes at room temperature. Wash cells twice with suspension buffer to remove residual fixative [11] [12].
- Permeabilization:
- For nuclear/transcription factor targets (e.g., NANOG): Resuspend the fixed cell pellet in 100 µL of 90% ice-cold methanol. Incubate for 15 minutes on ice. Wash with PBS [11] [12].
- For cytoplasmic targets or when using protein fluorophores: Resuspend in 100 µL of permeabilization buffer (e.g., 0.1% Triton X-100 or 0.1% Saponin in PBS). Incubate for 10-15 minutes at room temperature. Wash with a buffer containing the detergent if using saponin (as its effect is reversible) [11] [12].
- Simultaneous Intracellular & Surface Staining: Resuspend the fixed and permeabilized cells in a master mix containing antibodies against both intracellular and (if not applied in step 3) surface markers, diluted in an appropriate buffer. Incubate for 30 minutes in the dark at 4°C. This 2-step method (fix/perm followed by simultaneous staining) has been shown to be comparable to traditional serial staining, with the advantage of reduced cell loss and improved staining performance for some epitopes [14].
- Final Wash and Acquisition: Wash cells twice with wash buffer and resuspend in an appropriate volume for flow cytometry acquisition [13].
The Scientist's Toolkit: Essential Reagent Solutions
Table 2: Key Research Reagent Solutions for Intracellular Stem Cell Flow Cytometry
| Reagent Category | Specific Examples | Function & Rationale |
|---|---|---|
| Dissociation Agents | Accutase, Trypsin-EDTA, Collagenase | Generates a single-cell suspension from adherent cultures or tissues while preserving surface epitopes. |
| Fixatives | 4% Paraformaldehyde (PFA) [11] [12] | Cross-links proteins to preserve cellular structure and immobilize intracellular antigens. |
| Permeabilization Agents | Methanol [12], Triton X-100 [11] [12], Saponin [11] [12] | Disrupts lipid membranes to allow antibody access to intracellular compartments. Choice depends on target localization and antibody compatibility. |
| Blocking Agents | Goat Serum, Human IgG, FcR Blocking Reagents [11] | Binds to Fc receptors to prevent non-specific antibody binding, reducing background signal. |
| Viability Dyes | 7-AAD, DAPI [11] | Distinguishes live from dead cells during analysis; critical for excluding false positives from compromised cells. |
Advanced Analysis and Troubleshooting
Gating Strategies for Complex Stem Cell Populations
The analysis of multiparametric flow cytometry data requires a robust gating strategy. For stem cell populations, which are often rare, it is crucial to first gate on single cells using FSC-H vs FSC-A to exclude doublets, followed by gating on viable cells (using a viability dye), before finally analyzing marker expression [15]. When working with 10-color experiments, the use of fluorescence-minus-one (FMO) controls is essential to accurately set positive population gates and account for spectral overlap and data spread [15].
Troubleshooting Common Issues
The fixation and permeabilization method must be tailored to the specific stem cell marker and antibody being used.
Table 3: Troubleshooting Fixation and Permeabilization for Stem Cell Markers
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| High Background/Noise | Inadequate blocking; residual fixative; cell death. | Optimize FcR blocking; increase post-fixation washes; ensure high initial viability [11]. |
| Loss of Signal | Epitope damaged by fixative; incompatible permeabilization method. | Try alternative fixatives (e.g., acetone for some targets) [12]; switch permeabilization agent (e.g., from methanol to saponin) [12]. |
| Cell Clumping Post-Fixation | Excessive centrifugation force; inadequate resuspension. | Use gentle centrifugation (~200-300 x g) [11]; vortex gently during fixation steps [12]. |
| Poor Resolution of Dim Markers | Fluorochrome brightness not matched to antigen density. | Pair low-abundance intracellular targets (e.g., some transcription factors) with the brightest fluorochromes available [15]. |
The path to high-quality flow cytometry data for intracellular stem cell marker analysis is paved during the initial sample preparation. A high-viability, clump-free single-cell suspension is not merely a suggestion but an absolute requirement for generating reliable, reproducible, and publication-quality results. By adhering to the optimized protocols for dissociation, fixation, and permeabilization outlined in this application note, researchers can confidently proceed with multiparametric analysis, ensuring that the full potential of flow cytometry is realized in unraveling the complexities of stem cell biology.
Principles of Cell Fixation and Permeabilization for Accessing Intracellular Targets
The accurate assessment of intracellular proteins is a cornerstone of modern stem cell research, enabling scientists to define cellular identity, differentiation status, and functional state within heterogeneous populations. For human pluripotent stem cell (hPSC) derivatives, which are valuable for disease modeling, drug testing, and personalized medicine approaches, determining cell type identity in cultures is essential but challenging due to inherent heterogeneity and variations in differentiation efficiency among cell lines and protocols [16]. Flow cytometry offers a powerful solution for single-cell analysis of intracellular targets, but requires meticulous sample preparation to preserve cellular structure while allowing antibody access to internal epitopes.
This application note details the fundamental principles and optimized protocols for cell fixation and permeabilization specifically tailored for intracellular stem cell marker analysis. The methods outlined herein support the development of standardized approaches necessary for obtaining rigorous, reproducible data in stem cell research and drug development applications.
Core Principles of Fixation and Permeabilization
Strategic Purpose of Fixation
Fixation serves to preserve cellular architecture and stabilize protein targets for detection. By cross-linking proteins or precipitating cellular components, fixatives immobilize intracellular antigens while maintaining light scatter properties essential for flow cytometric analysis [10] [11]. The choice of fixative significantly impacts epitope preservation and must be optimized for specific intracellular targets.
Formaldehyde-based fixatives (typically 1-4% concentration) create reversible cross-links between proteins, preserving cellular morphology while maintaining accessibility for many intracellular epitopes. Methanol-free formaldehyde is recommended to prevent potential fluorescence quenching [17]. Organic solvent fixatives like methanol and acetone precipitate cellular components, often providing superior detection of certain intracellular targets, particularly cytoskeletal proteins and some transcription factors [11].
Strategic Purpose of Permeabilization
Permeabilization disrupts lipid membranes to enable antibody access to intracellular compartments. The choice of permeabilizing agent must be compatible with both the fixation method and the subcellular localization of the target antigen [11].
Mild detergents including saponin, Tween-20, and digitonin create small pores in membrane structures without complete dissolution, making them suitable for cytoplasmic antigens and soluble nuclear antigens [11]. Strong detergents such as Triton X-100 and NP-40 partially dissolve nuclear membranes, providing better access to nuclear antigens and some tightly-bound cytoplasmic proteins [11]. Organic solvents like methanol and acetone simultaneously fix and permeabilize cells through dehydration and lipid dissolution, but may destroy some epitopes [17].
Experimental Workflows
The following diagram illustrates the decision-making workflow for selecting appropriate fixation and permeabilization methods based on experimental requirements:
Method Selection Guide
Fixation and Permeabilization Method Comparison
Table 1: Comparison of Fixation and Permeabilization Methods for Intracellular Antigens
| Method | Mechanism of Action | Optimal Antigen Types | Advantages | Limitations |
|---|---|---|---|---|
| Aldehyde Fixation | Protein cross-linking via methylene bridges | Cell surface markers, many cytoplasmic proteins, membrane-associated proteins | Excellent morphology preservation, reversible cross-linking, compatible with most fluorophores | May mask some epitopes, requires permeabilization step, over-fixation can reduce antibody binding |
| Methanol Fixation | Protein precipitation and dehydration | Cytoskeletal proteins, viral antigens, some enzymes, transcription factors | Simultaneous fixation and permeabilization, excellent for nuclear antigens, enhances fluorescence intensity for some dyes | Alters light scatter properties, may destroy some epitopes, not suitable for all cell types |
| Acetone Fixation | Lipid dissolution and protein precipitation | Cytoskeletal elements, nuclear antigens, phosphorylated epitopes | Rapid action, simultaneous fixation and permeabilization, preserves enzyme activities | Complete dehydration, fragile cells may not withstand treatment, requires immediate processing |
| Saponin Permeabilization | Cholesterol complexation creating membrane pores | Cytoplasmic antigens, secretory granules, Golgi apparatus | Reversible process, gentle on epitopes, suitable for labile antigens | Temporary effect requiring continuous presence, may not access nuclear antigens effectively |
| Triton X-100 Permeabilization | Lipid solubilization and membrane dissolution | Nuclear antigens, cytoskeletal components, mitochondrial proteins | Strong permeabilization, permanent effect, suitable for difficult-to-access epitopes | Can disrupt protein complexes, may damage some epitopes, affects light scatter properties |
Research Reagent Solutions
Table 2: Essential Reagents for Intracellular Flow Cytometry
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Fixatives | 4% Formaldehyde (methanol-free) [16] [17], 100% Methanol [17], 100% Acetone [11] | Preserves cellular architecture and immobilizes antigens | Methanol-free formaldehyde prevents fluorescence quenching; organic solvents require chilling before use |
| Permeabilization Agents | Saponin [16], Triton X-100 [11], Tween-20 [11] | Disrupts lipid membranes to enable antibody access to intracellular compartments | Concentration optimization required (typically 0.1-1% in PBS); selection depends on antigen location |
| Buffers & Solutions | Flow Buffer (PBS + 0.5-1% BSA) [16] [11], Antibody Dilution Buffer [17], FcR Blocking Reagent [11] | Provides optimal staining environment, reduces non-specific binding | BSA concentration affects background staining; Fc receptor blocking essential for intracellular staining |
| Viability Dyes | 7-AAD, DAPI, TOPRO-3 [11], Propidium Iodide [18] | Distinguishes live from dead cells to exclude non-specific antibody binding | Must use amine-reactive fixable dyes if staining prior to fixation; choose dyes with non-overlapping emission spectra |
| Enzymatic Dissociation Reagents | Liberase-TH [16], Accutase [16], TrypLE [16] | Generates single-cell suspensions from adherent cultures or tissues | Gentle enzymes preserve surface and intracellular epitopes; optimization required for different stem cell types |
Detailed Experimental Protocols
Combined Surface and Intracellular Staining Protocol
The following protocol enables simultaneous detection of surface markers and intracellular antigens, particularly valuable for stem cell characterization where defining cellular identity requires multi-parameter analysis [19]:
Protocol Steps:
Sample Preparation: Harvest cells using gentle enzymatic dissociation (e.g., Liberase-TH for hPSC-derived cardiomyocytes or Accutase for undifferentiated hPSCs) to preserve epitope integrity [16]. Generate single-cell suspension and determine cell count and viability (should be >90%) [11].
Surface Antigen Staining: Resuspend 1×10^6 cells in 100 μL flow buffer containing titrated concentrations of fluorochrome-conjugated antibodies against surface markers. Incubate for 30-60 minutes at 4°C in the dark [19] [11].
Fixation: Pellet cells by centrifugation (200 × g for 5 minutes), remove supernatant, and resuspend in 100 μL of 4% methanol-free formaldehyde. Incubate for 15-20 minutes at room temperature [17].
Permeabilization: Wash cells twice with 3 mL PBS, then resuspend in permeabilization solution (e.g., 0.1% Triton X-100 or 0.5% saponin in PBS). Incubate for 10-15 minutes at room temperature [11].
Fc Receptor Blocking: Wash cells once with flow buffer, then resuspend in Fc blocking solution (2-10% serum matching secondary antibody host species or specific FcR blocking reagents). Incubate for 30-60 minutes at 4°C [11].
Intracellular Staining: Without washing, add titrated primary antibodies directly to blocking solution. Incubate for 60 minutes at room temperature or overnight at 4°C for low-abundance targets. Wash twice with permeabilization buffer [16] [19].
Secondary Antibody Detection (if needed): For unconjugated primary antibodies, resuspend cells in fluorochrome-conjugated secondary antibodies diluted in permeabilization buffer. Incubate for 30 minutes at room temperature in the dark. Wash twice with permeabilization buffer followed by one wash with standard flow buffer [17].
Data Acquisition: Resuspend cells in 200-500 μL PBS and analyze immediately on flow cytometer. Include appropriate controls: unstained cells, fluorescence minus one (FMO) controls, and isotype controls [18].
Methanol Fixation and Permeabilization Protocol
For targets that benefit from organic solvent treatment, such as transcription factors and some cytoskeletal proteins:
Cell Preparation: Generate single-cell suspension as described in Section 5.1. Pellet cells by centrifugation (150-300 × g for 5 minutes) and remove supernatant completely [17].
Fixation: Resuspend cells in approximately 100 μL of 4% formaldehyde per 1 million cells. Mix well to dissociate pellet and prevent cross-linking of individual cells. Fix for 15 minutes at room temperature [17].
Methanol Permeabilization: Permeabilize cells by adding ice-cold 100% methanol slowly to pre-chilled cells while gently vortexing, to a final concentration of 90% methanol. Alternatively, remove formaldehyde by centrifugation and resuspend in ice-cold 90% methanol (v/v in PBS) [17].
Storage or Immediate Use: Cells can be stored at -20°C in 90% methanol for several weeks or used immediately for immunostaining [17].
Immunostaining: Aliquot desired number of cells (generally 5×10^5 to 1×10^6 cells per assay). Wash cells by centrifugation in excess PBS to remove methanol. Resuspend cells in 100 μL of diluted primary antibody prepared in antibody dilution buffer. Incubate for 1 hour at room temperature. Continue with washing and secondary antibody detection as needed [17].
Critical Parameters for Success
Antibody Validation and Titration
The specificity of an antibody is always context-dependent, requiring "fit-for-purpose" validation for each application [16]. Proper antibody titration is essential for optimal signal-to-noise ratio and must be performed for each new antibody lot and cell type. Validation should include:
- Demonstration of specific staining pattern consistent with expected subcellular localization
- Loss of signal in knockout cells or with competing immunogen
- Comparison with well-characterized positive and negative control cell types [16]
Appropriate Control Strategies
Comprehensive controls are mandatory for accurate interpretation of intracellular flow cytometry data:
- Biological controls: Include known positive and negative cell populations (e.g., undifferentiated hPSCs as negatives for cardiomyocyte markers) [16]
- Isotype controls: Match the host species, isotype, and conjugation of primary antibodies
- Fluorescence Minus One (FMO) controls: Essential for polychromatic panels to establish gating boundaries [18]
- Unstained cells: Assess autofluorescence levels and instrument settings
- Compensation controls: Required for multicolor experiments, using compensation beads or stained cells [18]
Data Acquisition and Analysis Standards
When publishing flow cytometry data, include comprehensive methodological details:
- Instrument manufacturer, model, and software versions [18]
- Laser lines and optical emission filters used [18]
- Number of events collected and gating strategy [18]
- Compensation methodology and software used for analysis [18]
- Detailed sample preparation procedures including all proteases, fixatives, and permeabilization reagents [18]
Applications in Stem Cell Research
The protocols described herein enable critical applications in stem cell research and drug development:
- Assessment of differentiation efficiency in hPSC derivatives through intracellular marker expression (e.g., cardiac troponin in cardiomyocytes) [16]
- Characterization of rare stem cell populations through combined surface and intracellular marker analysis [10] [19]
- Identification of novel surface marker combinations that correlate with intracellular lineage markers, enabling live cell sorting without fixation [19]
- Cell cycle analysis of stem cell populations through DNA content quantification [10]
- Analysis of signaling pathway activation through intracellular phospho-protein detection [11]
When properly optimized and validated, these fixation and permeabilization approaches provide the foundation for robust, reproducible intracellular analysis that advances stem cell research and therapeutic applications.
Stem cell research represents a frontier in developmental biology and regenerative medicine, but its progress is inherently linked to our ability to accurately identify and characterize stem cell populations. The primary challenges in this field stem from the heterogeneous nature of stem cell populations and the lack of standardized protocols across laboratories. Flow cytometry emerges as a powerful solution, offering single-cell resolution and multiparametric analysis to navigate these complexities [10]. This application note details optimized methodologies for stem cell characterization, with particular emphasis on intracellular marker detection—a critical requirement for assessing pluripotency and differentiation status.
Technical Challenges in Stem Cell Analysis
Cellular Heterogeneity
Even within clonal stem cell populations, functional heterogeneity exists due to variations in gene expression, cell cycle status, and spontaneous differentiation. This heterogeneity complicates analysis using bulk techniques like Western blotting or qRT-PCR, which provide population averages and mask important minority subpopulations [10]. Flow cytometry addresses this limitation by enabling:
- Single-cell analysis of thousands to millions of individual cells
- Identification and quantification of rare cell populations (e.g., residual undifferentiated cells in differentiated cultures)
- Simultaneous assessment of multiple stem cell markers alongside functional parameters
Standardization Issues
The lack of standardized protocols across research facilities presents a significant challenge, particularly when comparing results between laboratories or establishing clinical-grade stem cell lines [10]. Key standardization challenges include:
- Variability in sample preparation methods (dissociation techniques, fixation protocols)
- Inconsistencies in antibody selection and titration
- Divergent gating strategies and data analysis approaches
- Inadequate validation of antibody specificity for stem cell markers
Essential Stem Cell Markers for Flow Cytometry
The identification of stem cells relies on specific molecular signatures comprising both surface and intracellular markers. The table below summarizes key markers utilized for characterizing various stem cell types.
Table 1: Essential Markers for Stem Cell Characterization
| Marker Type | Specific Markers | Stem Cell Population | Localization | Biological Function |
|---|---|---|---|---|
| Pluripotency Markers | SSEA-4, TRA-1-60, TRA-1-81 | Human embryonic stem cells (hESCs), induced pluripotent stem cells (iPSCs) | Cell Surface | Maintain undifferentiated state; indicators of pluripotency |
| Pluripotency Markers | Nanog, Oct-3/4, Sox2 | hESCs, iPSCs | Intracellular (Nuclear) | Core pluripotency transcription factors; regulate self-renewal |
| Hematopoietic Stem Cell Markers | CD34, CD133, CD90 | Hematopoietic stem cells (HSCs) | Cell Surface | Cell adhesion, progenitor cell identification |
| Mesenchymal Stem Cell Markers | CD73, CD90, CD105 | Mesenchymal stem cells (MSCs) | Cell Surface | Immunomodulation, tissue repair capacity |
| Neural Stem Cell Markers | Nestin, Sox2, Musashi-1 | Neural stem cells (NSCs) | Intracellular | Cytoskeletal protein; maintains neural progenitor state |
Optimized Protocol for Intracellular Stem Cell Marker Analysis
Stage 1: Sample Preparation and Viability Assessment
Purpose: Generate high-quality single-cell suspensions while preserving cell viability and antigen integrity.
Materials:
- Appropriate cell culture medium
- Enzyme-free cell dissociation buffer (for sensitive epitopes)
- Suspension/wash buffer (PBS with 5-10% fetal calf serum)
- Viability dye (7-AAD, DAPI, or fixable amine-reactive dyes)
- Polystyrene round-bottom tubes (12 × 75 mm)
Procedure:
- Gently harvest cells using minimal mechanical disruption to prevent clumping
- For adherent cultures, prefer enzyme-free dissociation methods when possible to preserve surface epitopes
- Wash cells by centrifugation at 200 × g for 5 minutes at 4°C
- Resuspend cell pellet in ice-cold suspension buffer at recommended density (0.5-1 × 10^6 cells/mL)
- Stain with appropriate viability dye according to manufacturer's protocol
- Perform two wash steps with wash buffer (200 × g, 5 minutes, 4°C) [11]
Critical Considerations:
- Maintain cells at 4°C throughout the procedure to prevent internalization of surface markers
- Avoid excessive centrifugation force or vortexing to prevent cell damage
- Determine cell count and viability (should be ≥90% for optimal results)
- Include unstained and single-stained controls for compensation
Stage 2: Surface Antigen Staining
Purpose: Identify and characterize stem cell populations based on surface marker expression before intracellular staining.
Procedure:
- Resuspend cell pellet in cold suspension buffer
- Add fluorochrome-conjugated antibodies against surface markers (SSEA-4, TRA-1-60, CD34, etc.)
- Incubate in the dark for 30 minutes at 4°C
- Wash twice with cold suspension buffer (200 × g, 5 minutes, 4°C)
- Proceed to fixation or analyze immediately for surface markers only [11]
Stage 3: Fixation and Permeabilization
Purpose: Preserve cellular architecture while allowing antibody access to intracellular targets.
Materials:
- Fixative (1-4% paraformaldehyde, 90% methanol, or acetone)
- Permeabilization solution (Triton X-100, NP-40, Tween 20, or saponin)
- Permeabilization wash buffer
Procedure:
- Fix cells by resuspending pellet in appropriate fixative:
- 1-4% PFA: 15-20 minutes on ice
- 90% methanol: 10 minutes at -20°C
- Acetone: 10-15 minutes on ice (not compatible with plastic tubes)
- Wash cells twice with suspension buffer
- Permeabilize cells by incubating with detergent solution:
- Harsh detergents (Triton X-100, NP-40 at 0.1-1%): 10-15 minutes at room temperature for nuclear antigens
- Mild detergents (Tween 20, saponin at 0.2-0.5%): 10-15 minutes at room temperature for cytoplasmic antigens
- Wash twice with permeabilization wash buffer [11]
Critical Considerations:
- Acetone fixation also permeabilizes cells; no additional permeabilization step required
- Methanol fixation can destroy some epitopes; test multiple fixatives for new targets
- Optimal detergent concentration should be determined empirically for each antigen
Stage 4: Intracellular Staining
Purpose: Detect and quantify intracellular stem cell markers (transcription factors, cytoplasmic proteins).
Procedure:
- Resuspend fixed and permeabilized cells in permeabilization buffer
- Add fluorochrome-conjugated antibodies against intracellular targets (Nanog, Oct-3/4, Sox2, etc.)
- Incubate in the dark for 30-60 minutes at 4°C (or according to antibody manufacturer's recommendation)
- Wash twice with permeabilization buffer
- Resuspend in suspension buffer for flow cytometry acquisition [11] [13]
Critical Considerations:
- Include isotype controls for intracellular targets to assess non-specific binding
- Titrate all antibodies to determine optimal signal-to-noise ratio
- For nuclear targets (transcription factors), ensure adequate permeabilization of nuclear membrane
Data Analysis and Gating Strategy
Proper data analysis is crucial for accurate interpretation of stem cell heterogeneity. The workflow below outlines a systematic approach to identify and characterize stem cell populations.
Diagram 1: Gating Strategy for Stem Cell Analysis
Quantitative Analysis of Heterogeneous Populations
When analyzing stem cell populations, particularly after differentiation protocols, it's essential to accurately quantify subpopulations:
Back-gating calculations: When analyzing nested populations, calculate the percentage of the total population
- Example: If 30.1% of total cells are target stem cells, and 14.5% of these express a specific intracellular marker, then 4.36% (30.1 × 0.145) of the total sample are marker-positive stem cells [20]
Mean Fluorescence Intensity (MFI): Provides a relative measure of antigen abundance, useful for assessing differentiation status
Contour plots versus dot plots: Use contour plots to better visualize small populations that might be overlooked in standard dot plots [21]
Research Reagent Solutions
The table below outlines essential reagents and their specific functions in stem cell flow cytometry protocols.
Table 2: Essential Research Reagents for Stem Cell Flow Cytometry
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Viability Dyes | 7-AAD, DAPI, TOPRO-3 | Distinguish live/dead cells based on membrane integrity | DNA-binding dyes; cannot use with fixed cells [11] |
| Fixable Viability Dyes | Amine-reactive dyes | Covalently bind to amines in dead cells | Compatible with fixation/permeabilization |
| Fixation Reagents | 1-4% Paraformaldehyde, 90% Methanol, Acetone | Preserve cellular structure and antigen availability | Methanol may destroy some epitopes; acetone also permeabilizes [11] |
| Permeabilization Detergents | Triton X-100, NP-40, Saponin, Tween 20 | Disrupt membranes for antibody internalization | Harsh detergents (Triton) for nuclear antigens; mild for cytoplasmic [11] |
| Blocking Reagents | Goat serum, Human IgG, Mouse anti-CD16/CD32 | Prevent non-specific antibody binding | Critical for reducing background; species-specific |
| Antibody Panels | Fluorochrome-conjugated antibodies against stem cell markers | Detect specific surface and intracellular antigens | Include pluripotency, differentiation, and lineage markers [13] |
Advanced Applications and Future Directions
Imaging Flow Cytometry
Imaging flow cytometry (IFC) combines the high-throughput capability of conventional flow cytometry with morphological analysis, providing:
- Spatial information about subcellular localization of stem cell markers
- Morphometric parameters for distinguishing stem cell states
- Validation of staining specificity through visual confirmation [10]
Organoid Analysis
Flow cytometry is increasingly applied to characterize complex 3D stem cell-derived models:
- Dissociation protocols for organoids into single-cell suspensions
- Identification of multiple cell types within the same organoid
- Assessment of cellular heterogeneity across different organoid batches [10]
Standardization Initiatives
Efforts to address standardization challenges include:
- Development of validated antibody panels for specific stem cell types
- Protocol harmonization across research facilities using uniform antibody batches and methodologies
- Reference standards for instrument calibration and cross-laboratory comparisons [10]
Navigating the challenges of cell heterogeneity and standardization in stem cell analysis requires robust, reproducible methodologies. The flow cytometry protocols detailed in this application note provide a framework for reliable intracellular stem cell marker analysis. By implementing these optimized procedures—from sample preparation through data analysis—researchers can achieve more consistent results, better characterize stem cell populations, and advance the field toward greater standardization. As flow cytometry technologies continue to evolve, with innovations in imaging flow cytometry and high-parameter panels, our ability to resolve the complexities of stem cell biology will correspondingly enhance, accelerating progress in regenerative medicine and therapeutic development.
A Step-by-Step Optimized Protocol for Intracellular Staining of Stem Cells
The analysis of intracellular stem cell markers via flow cytometry is a cornerstone of modern regenerative medicine and drug development research. The fidelity of this analysis is entirely dependent on the initial quality of the single-cell suspension. Human induced pluripotent stem cells (hiPSCs), characterized by their unlimited self-renewal and capability to differentiate into all three germ layers, are particularly fragile and prone to apoptosis upon dissociation [22]. Therefore, a meticulous approach to their culture, harvesting, and preparation is non-negotiable for obtaining meaningful flow cytometry data. This application note provides a detailed, step-by-step protocol for generating high-quality single-cell suspensions from hiPSCs, optimized specifically for subsequent intracellular staining and flow cytometric analysis within a research setting.
Strategic Planning and Reagent Selection
Successful preparation of hiPSCs for flow cytometry begins with strategic planning and the use of defined reagents. The choice of culture system and dissociation method is critical to maintain cell viability, pluripotency, and to minimize spontaneous differentiation.
Key Research Reagent Solutions
The following table summarizes the essential reagents and their functions for the effective culture and preparation of hiPSCs.
Table 1: Essential Reagents for hiPSC Culture and Single-Cell Suspension Preparation
| Reagent Category | Specific Examples | Function | Key Considerations |
|---|---|---|---|
| Defined Culture Medium | Essential 8 (E8) Medium, mTeSR Plus [22] [23] | Supports hiPSC growth and expansion under chemically defined, feeder-free conditions. | Simpler formulation than earlier media; requires daily changes. |
| Coatings/Matrices | GFR Matrigel, Vitronectin XF, Laminin-521 [22] | Mimics the extracellular matrix to aid cell attachment and expansion. | Critical for feeder-free culture. GFR Matrigel is "growth factor reduced" for better control. |
| Non-Enzymatic Dissociation Agent | Versene Solution (EDTA) [22] | Gently dissociates cells by chelating calcium and magnesium, preventing enzyme-induced damage. | Preferred for routine passaging; improves cell survival and replating efficiency. |
| Enzymatic Dissociation Agent | Accutase [23] | A mixture of enzymes that dissociates cells into a single-cell suspension. | Used for applications requiring a complete single-cell suspension, such as flow cytometry preparation. |
| Rho-associated Kinase (ROCK) Inhibitor | Y-27632 [23] | Promotes cell survival and inhibits apoptosis following single-cell dissociation. | Typically added to the medium for 24 hours after passaging or thawing. |
Detailed Experimental Protocols
Protocol 1: Culturing and Maintaining hiPSCs
Principle: To maintain hiPSCs in a pristine, undifferentiated state, ready for experimentation. This protocol uses a feeder-free system with a chemically defined medium [22].
Materials:
- Coated tissue culture vessel (e.g., 6-well plate coated with GFR Matrigel)
- Essential 8 (E8) Medium or mTeSR Plus medium
- DPBS (without calcium and magnesium)
- Versene solution
- 37°C incubator with 5% CO₂
Procedure:
- Coating: Thaw GFR Matrigel on ice and dilute in cold DMEM/F-12. Coat the culture vessel surface and incubate at room temperature for at least 1 hour.
- Feeding: Aspirate the coating solution and add pre-warmed E8 medium. Return the culture to the incubator.
- Daily Monitoring and Feeding: Observe hiPSC colonies daily under a microscope. Check for a high nucleus-to-cytoplasm ratio, distinct colony edges, and the absence of spontaneously differentiated cells. Change the medium daily.
- Passaging (at ~80% confluence): a. Aspirate the medium and gently wash the cells with DPBS. b. Add enough Versene solution to cover the surface and incubate at 37°C for 3-5 minutes. c. Monitor cells under the microscope. When colony edges begin to detract, carefully aspirate the Versene. d. Add fresh E8 medium and gently scrape the cells off the surface using a pipette tip or cell scraper. e. Break the cell clusters into smaller clumps (100-1000 cells) by pipetting gently 2-3 times. f. Transfer the cell suspension to a new coated vessel at the desired split ratio (typically 1:6 to 1:12).
- Quality Control: Regularly inspect cultures for spontaneous differentiation. Differentiated areas can be manually removed before passaging.
Protocol 2: Harvesting and Creating a Single-Cell Suspension
Principle: To generate a robust, high-viability single-cell suspension from cultured hiPSCs suitable for flow cytometry analysis. This protocol employs enzymatic dissociation and incorporates a survival additive [23] [24].
Materials:
- hiPSC culture at ~80% confluence
- DPBS
- Accutase enzyme cell detachment medium
- Flow Cytometry Staining Buffer (PBS with 0.5-1% BSA)
- Rho-associated kinase (ROCK) inhibitor (Y-27632)
- Centrifuge and 15 mL conical tubes
Procedure:
- Preparation: Pre-warm Accutase to 37°C. Add ROCK inhibitor (e.g., 10 µM Y-27632) to the necessary volume of fresh culture medium for the subsequent step [23].
- Wash: Aspirate the culture medium from the hiPSCs and gently wash the cell layer with DPBS to remove residual serum and calcium.
- Dissociation: Add enough Accutase to cover the cell layer. Incubate at 37°C for 5-10 minutes. Monitor under the microscope until >90% of cells are detached and rounded up.
- Neutralization and Collection: Gently pipette the Accutase over the surface to aid detachment. Transfer the cell suspension to a conical tube containing an equal volume of culture medium with ROCK inhibitor to neutralize the enzyme.
- Wash and Count: Centrifuge the cell suspension at 300-400 x g for 4-5 minutes. Carefully decant the supernatant. Resuspend the cell pellet in Flow Cytometry Staining Buffer and perform a cell count and viability analysis (e.g., using Trypan Blue exclusion).
- Final Preparation for Flow Cytometry: Centrifuge again and resuspend the cell pellet in an appropriate volume of Flow Cytometry Staining Buffer to a final concentration of 1 x 10⁷ cells/mL [24]. The cell suspension is now ready for subsequent staining procedures.
The following workflow diagram summarizes the key stages of the complete process from culture to suspension preparation.
Critical Parameters for High-Quality Suspensions
Table 2: Key Parameters for Optimal Single-Cell Suspension Quality
| Parameter | Target | Rationale |
|---|---|---|
| Cell Viability | >90% | Dead cells increase background noise, bind antibodies non-specifically, and can clog the flow cytometer. |
| Single-Cell State | >95% single cells, minimal doublets/clumps | Cell clogs can clog the instrument's fluidics system and result in inaccurate, multi-cell readings. |
| Final Cell Concentration | ~1 x 10⁷ cells/mL [24] | An optimal concentration ensures an event rate that the flow cytometer can process efficiently without coincidence. |
| Use of ROCK Inhibitor | 10 µM Y-27632 during/after dissociation [23] | Dramatically improves survival of hiPSCs after enzymatic dissociation into single cells. |
| Sterility and Purity | Aseptic technique, no microbial contamination | Preserves cell health and prevents the introduction of contaminants that can interfere with analysis. |
Troubleshooting Common Issues
- Poor Cell Viability After Harvest: Ensure ROCK inhibitor is added to the recovery medium. Avoid over-incubation with Accutase. Gently pipette during neutralization, and do not vortex.
- Excessive Cell Clumping: Filter the cell suspension through a sterile cell strainer (e.g., 40 µm nylon mesh) before analysis. Ensure complete enzymatic dissociation and avoid excessive force when pipetting.
- Spontaneous Differentiation in Culture: Do not let cultures become over-confluent. Change medium daily without fail. Meticulously remove any differentiated areas before passaging.
The journey to reliable flow cytometry data for intracellular stem cell markers begins at the bench long before the sample reaches the instrument. Adherence to the detailed protocols outlined here—utilizing defined culture systems, gentle enzymatic dissociation with pro-survival additives, and rigorous attention to cell handling—will consistently yield high-quality single-cell suspensions from hiPSCs. This foundational step is critical for the accurate assessment of pluripotency, the validation of stem cell quality, and the successful execution of downstream applications in disease modeling and drug development.
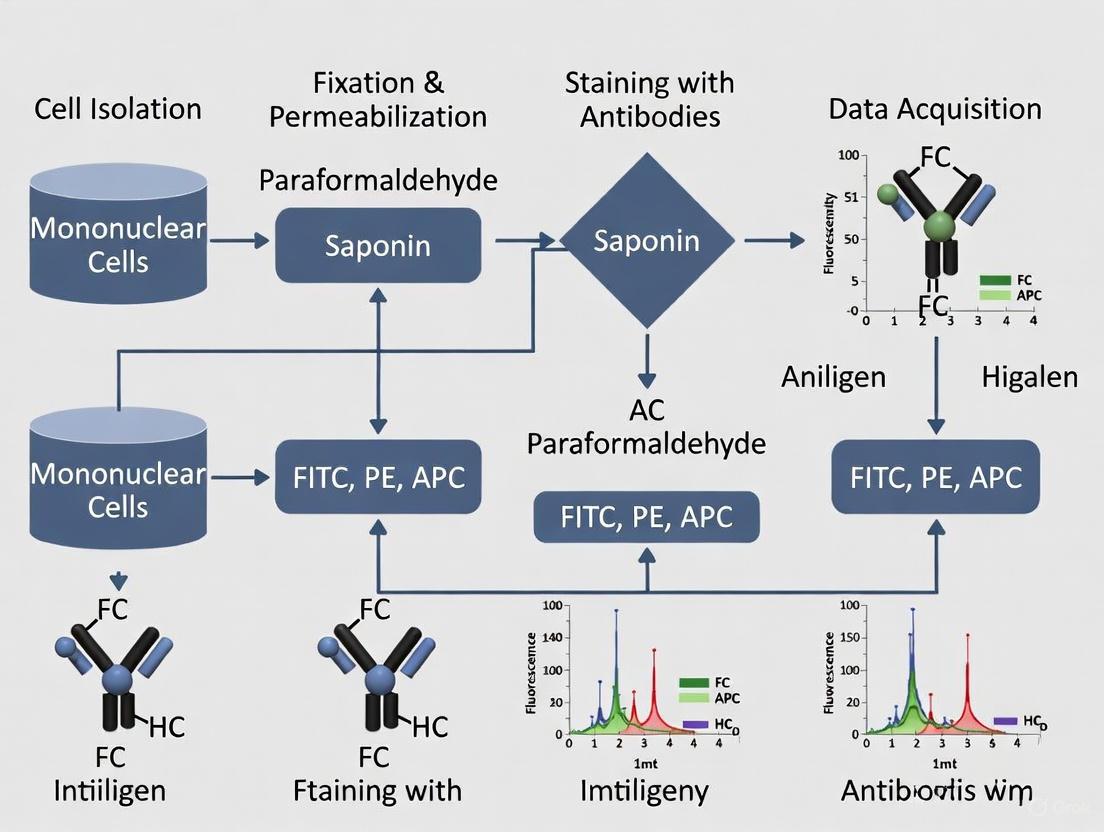
The accurate detection of nuclear transcription factors by flow cytometry is a critical tool in stem cell research, enabling scientists to dissect the complex regulatory networks that govern pluripotency, self-renewal, and differentiation. Transcription factors such as Nanog, Sox2, and Oct4 are pivotal in maintaining stem cell identity, and their precise intracellular measurement is essential for characterizing stem cell populations and optimizing differentiation protocols [7]. Success in these assays hinges on the rigorous optimization of fixation and permeabilization (Fix/Perm) methods. These steps must be sufficiently robust to allow antibodies access to the nucleus while preserving epitope integrity and cellular morphology. This application note provides a detailed framework for selecting the appropriate reagents and methods to ensure the reliable and reproducible staining of nuclear transcription factors, framed within the broader context of intracellular stem cell marker research.
The Critical Role of Fixation and Permeabilization
Unlike cell surface markers or cytoplasmic proteins, nuclear transcription factors present a unique challenge. They are often localized within the nucleus and can be bound to DNA or exist within complex protein assemblies [7]. The primary goal of fixation is to stabilize the cell's structure and prevent the loss of intracellular contents. However, the cross-linking nature of many fixatives can sometimes mask the very epitopes that antibodies need to bind. Consequently, permeabilization—the process of creating holes in the lipid membranes—must be strong enough to allow large antibody-fluorophore complexes to traverse both the plasma and nuclear membranes to reach their targets [25].
The choice of Fix/Perm method is a balancing act. Gentle detergent-based systems, while excellent for many cytoplasmic targets, often provide insufficient permeabilization for nuclear antigens. Conversely, harsh alcohol-based methods can denature proteins and destroy the antigenicity of some transcription factors and many cell surface markers [7]. For nuclear transcription factors, specialized buffer systems that combine formaldehyde-based fixation with stronger permeabilizing agents are typically required to achieve optimal results [26] [27].
Selecting the Appropriate Buffer Systems
The selection of a Fix/Perm buffer system should be guided by the specific intracellular target. Commercial kits are often optimized for particular protein classes, providing a reliable starting point for assay development. The table below summarizes the primary buffer systems used for intracellular staining, with a focus on nuclear transcription factors.
Table 1: Comparison of Intracellular Staining Buffer Systems
| Buffer System Type | Primary Application | Key Characteristics | Example Products |
|---|---|---|---|
| Transcription Factor Buffers | Nuclear proteins (e.g., FoxP3, Sox17), Transcription factors | Combines formaldehyde fixation with strong permeabilization agents; necessary for nuclear membrane penetration and disrupting DNA/protein complexes [7]. | Foxp3/Transcription Factor Staining Buffer Set [26], BD Pharmingen Transcription Factor Buffer Set [7], Proteintech Foxp3 / Transcription Factor Staining Buffer Kit [27] |
| Cytokine Buffers | Cytoplasmic proteins (e.g., cytokines), Secreted proteins | Uses mild detergents like saponin; permeabilization is reversible, requiring antibodies to be diluted in permeabilization buffer [26] [7]. | Intracellular Fixation & Permeabilization Buffer Set [26], BD Cytofix/Cytoperm [7] |
| Methanol-Based Protocols | Phosphorylated signaling proteins (e.g., MAPK, STAT) | Involves formaldehyde fixation followed by ice-cold methanol permeabilization; a harsh process that can destroy some epitopes and surface markers but is optimal for many phospho-proteins [26] [28]. | N/A |
For researchers requiring the simultaneous detection of transcription factors and fluorescent proteins (e.g., GFP in reporter cell lines), a novel "Dish Soap Protocol" has been recently developed. This method uses a buffer containing common dishwashing detergent (e.g., Fairy/Dawn) to achieve a balance between sufficient permeabilization for nuclear access and the preservation of fluorescent protein signal, overcoming the limitations of many commercial kits [25].
Recommended Protocol for Nuclear Transcription Factors
The following step-by-step protocol is optimized for the staining of nuclear transcription factors in a 96-well plate format, incorporating best practices from major manufacturers and recent scientific literature [26] [27].
Materials and Reagents
Table 2: Research Reagent Solutions for Transcription Factor Staining
| Reagent | Function | Example Product / Composition |
|---|---|---|
| Foxp3/Transcription Factor Buffer Set | Specialized fixative and permeabilization buffers for nuclear antigens. | Contains Fixation/Permeabilization Concentrate and Diluent, plus 10X Permeabilization Buffer [26] [27]. |
| Flow Cytometry Staining Buffer | Wash and antibody resuspension buffer; contains protein to reduce background. | Ready-to-use buffer containing protein stabilizers [26]. |
| Fc Receptor Blocking Reagent | Blocks non-specific antibody binding to Fc receptors on immune cells. | Normal serum from the host species of the staining antibodies [26] [29]. |
| Fixable Viability Dye | Distinguishes live from dead cells; crucial as fixed dead cells exhibit high non-specific binding. | eFluor series dyes [26]. |
| Fluorochrome-Conjugated Antibodies | Detection of surface and intracellular targets. | Target-specific antibodies, titrated for optimal signal-to-noise. |
Staining Procedure
- Cell Preparation: Harvest and wash cells to create a single-cell suspension. Aliquot 0.5-1 x 10^6 cells per well in a 96-well U-bottom plate [27].
- Viability Staining (Optional but Recommended): Resuspend the cell pellet in a fixable viability dye diluted in PBS. Incubate for 10-30 minutes on ice, protected from light. Wash with an excess of staining buffer [26].
- Cell Surface Staining: Resuspend cells in staining buffer containing pre-titrated antibodies against cell surface markers (e.g., CD184 for definitive endoderm [7]). Incubate for 20-30 minutes on ice or at 4°C in the dark. Wash twice with staining buffer.
- Fixation and Permeabilization:
- Discard the supernatant after the final surface stain wash.
- Fix the cells by resuspending the pellet in 200 µL of freshly prepared 1X Foxp3 Fixation/Permeabilization working solution. Vortex briefly and incubate for 30-60 minutes at 4°C in the dark [26] [27].
- Wash cells with 200 µL of 1X Permeabilization Buffer. Centrifuge and discard the supernatant.
- Intracellular Staining: Resuspend the cell pellet in 100 µL of 1X Permeabilization Buffer. Add the recommended amount of directly conjugated antibody against the intracellular nuclear antigen (e.g., FoxP3, Sox17). Incubate for 30-60 minutes at room temperature in the dark.
- Final Washes and Acquisition: Wash the cells twice with 200 µL of 1X Permeabilization Buffer. Resuspend the final cell pellet in an appropriate volume of Flow Cytometry Staining Buffer. Analyze samples on a flow cytometer promptly [26].
The following workflow diagram outlines the key decision points and steps in this protocol.
Troubleshooting and Best Practices
- Simultaneous Staining with Fluorescent Proteins: For stem cell research using GFP or other fluorescent protein reporters, standard transcription factor buffers may quench the signal. The recently published "Dish Soap Protocol," which uses a fixative containing formaldehyde, Tween-20, and a small amount of dishwashing detergent (Fairy/Dawn), followed by permeabilization with a dilute dish soap solution, has been shown to effectively preserve GFP while allowing robust Foxp3 and other transcription factor staining [25].
- Epitope Sensitivity: The fixation and permeabilization steps can compromise the detection of some cell surface markers and intracellular epitopes. It is critical to validate the compatibility of all antibodies in your panel with the chosen Fix/Perm protocol [7] [25].
- Controls are Essential: Always include appropriate controls, such as fluorescence-minus-one (FMO) and isotype controls, to accurately set positive staining gates and identify non-specific background [18].
- Data Reporting: When publishing, provide a detailed description of the fixation and permeabilization reagents, including vendors, catalog numbers, and incubation times, to ensure the reproducibility of your findings [18].
The rigorous characterization of stem cell populations via nuclear transcription factor analysis demands meticulous attention to fixation and permeabilization techniques. By selecting buffer systems specifically designed for nuclear antigen detection, such as the Foxp3/Transcription Factor buffer sets, and following optimized staining protocols, researchers can generate high-quality, reproducible data. As the field advances, novel methods like the dish soap protocol offer new avenues for multiplexed analysis, further empowering discovery in stem cell biology and therapeutic development.
In high-parameter flow cytometry, the exquisite specificity of antibody binding is paramount for accurate measurement of proteins and other molecules at the single-cell level. However, this specificity can be compromised by various non-specific interactions that increase background noise and reduce assay sensitivity. For researchers investigating intracellular stem cell markers, where target proteins may be scarce and population resolution critical, implementing robust blocking strategies is essential for data integrity. Non-specific binding arises primarily through three mechanisms: Fc receptor-mediated antibody binding, low-affinity interactions with cellular components, and direct interactions between fluorophores and off-target cellular structures [30] [31]. Judicious application of blocking reagents significantly improves the signal-to-noise ratio by reducing these unwanted interactions, thereby enhancing the sensitivity needed to detect authentic biological signals above assay noise [30]. This application note provides a comprehensive framework for blocking strategy implementation within the context of intracellular stem cell marker analysis, featuring optimized protocols, reagent selection guidelines, and practical troubleshooting advice.
The Science of Non-Specific Binding in Flow Cytometry
Mechanisms of Undesirable Binding
The primary sources of non-specific binding in flow cytometry stem from well-characterized molecular interactions. Fc receptors (FcRs) expressed on various cell types, particularly within the hematopoietic system, can bind the constant region (Fc) of antibodies independent of their antigen-specific variable regions [30] [31]. This interaction is especially problematic when working with phagocytic cells like monocytes and macrophages, but can affect numerous cell types including B cells, dendritic cells, neutrophils, NK cells, and others [31]. The affinity of these interactions varies, with high-affinity receptors like CD64 (FcγRI) particularly impactful in high-parameter flow cytometry assays [30].
Beyond Fc-mediated binding, antibodies can engage in low-affinity interactions with off-target epitopes, especially when used at non-optimal concentrations [31]. Perhaps more insidiously, certain fluorophore classes can directly bind to cellular components. Brilliant dyes, NovaFluors, and Qdots are prone to dye-dye interactions, while tandems can break down and produce erroneous signals in channels corresponding to their constituent fluorophores [30]. Specific fluorochrome-cell interactions have been documented, such as the binding of PE-Cy5 conjugates to cells expressing certain receptors, and more recently, evidence of antibodies themselves binding to specific fluorochromes like AlexaFluor 700 [31]. For stem cell researchers, these issues are compounded when analyzing intracellular markers, as permeabilization exposes a much larger range of potential off-target epitopes [30].
Impact on Stem Cell Research
For scientists working with induced pluripotent stem cells (iPSCs) and other stem cell populations, accurate measurement of undifferentiated stem cell markers is crucial for defining pluripotent status and evaluating differentiation capacity [13]. The line-to-line variability in differentiation potential observed in iPSCs makes verification of pluripotent status through marker expression particularly important [13]. When non-specific binding inflates background fluorescence, it becomes challenging to distinguish between truly positive populations and negative or low-expressing populations, potentially leading to misinterpretation of stem cell characterization data. Furthermore, the high-value nature of these cellular models demands protocols that maximize data quality from often-limited sample sizes.
Strategic Blocking Approaches
Fc Receptor Blocking
Blocking Fc-mediated binding represents the most fundamental step in reducing non-specific signal. The strategic approach to Fc blocking depends on both the host species of the staining antibodies and the species origin of the cells being analyzed [30]. The general principle is to use normal sera or purified immunoglobulins from the same species as the primary antibodies being used for staining [30] [31]. For example, when staining mouse cells with predominantly rat antibodies, optimal blocking is achieved with normal rat serum [30] [31]. Conversely, for human targets stained with mouse antibodies, which bind well to human FcγR, normal mouse serum or purified mouse IgG is recommended [30] [31].
Research comparing blocking reagents has demonstrated that purified human IgG effectively reduces non-specific binding of isotype controls to background fluorescence levels in human monocyte-derived macrophages [31]. This approach offers advantages over normal serum, including reduced lot-to-lot variation and elimination of components that might inadvertently activate cells [31]. Commercial FcR blocking reagents containing antibodies against specific Fc receptors (e.g., anti-CD16/CD32) provide a more targeted approach and are particularly effective for specific cell types [11] [9] [31].
Table 1: Fc Receptor Blocking Reagent Selection Guide
| Cell Type | Antibody Host | Recommended Blocking Reagent | Alternative Options |
|---|---|---|---|
| Mouse cells | Rat monoclonal | Normal rat serum [31] | Purified rat IgG, Fc Block (anti-CD16/32) [9] [31] |
| Human cells | Mouse monoclonal | Purified mouse IgG [31] | Normal mouse serum [31], Human IgG [32], Fc Block (anti-CD16/32) [9] |
| Multiple species | Mixed host | Combination sera from antibody host species [30] | Species-specific FcR blocking antibodies |
Fluorophore and Dye Interaction Blocking
With the proliferation of novel fluorochromes in high-parameter panels, blocking dye-specific interactions has become increasingly important. Certain dye classes, particularly cyanine-based tandems and polymer dyes like Brilliant Violet and Brilliant Ultra Violet, are prone to non-specific interactions [30] [31]. These interactions can occur between dyes themselves or between dyes and cellular components, leading to erroneous signals that can be misassigned to different markers during analysis [30].
Specialized blocking reagents have been developed to address these challenges. For panels containing SIRIGEN "Brilliant" or "Super Bright" polymer dyes, Brilliant Stain Buffer is essential to prevent dye-dye interactions [30]. The polyethylene glycol (PEG) in this buffer also reduces non-specific binding of many non-Brilliant fluorophores, particularly relevant for samples from donors immunized with PEG-containing vaccines [30]. For other specific fluorochrome interactions, reagents such as True-Stain Blocker have demonstrated efficacy in minimizing binding to monocytes [31]. Additionally, "Oligo-Block" (phosphorothioate-oligodeoxynucleotides) has been shown to effectively block cyanine-tandem binding to human monocytes [31].
Table 2: Fluorophore-Specific Blocking Reagents
| Fluorophore Class | Blocking Reagent | Mechanism | Application Notes |
|---|---|---|---|
| Brilliant dyes (SIRIGEN polymers) | Brilliant Stain Buffer / Brilliant Stain Buffer Plus [30] | Prevents polymer dye-dye interactions | Use at up to 30% (v/v) in staining mix; Plus version offers 4x reduction in volume [30] |
| Cyanine tandems (Cy5, Cy5.5, etc.) | Oligo-Block [31] | Blocks charge-mediated binding | Effective for monocytes; particularly relevant for PE-Cy5 and similar tandems [31] |
| Multiple dye classes | True-Stain Blocker [31] | Reduces non-specific fluorophore binding | Validated on human monocytes; useful for various fluorochrome types [31] |
| NovaFluors | CellBlox [30] | Prevents dye-specific interactions | Required for panels containing NovaFluors [30] |
Comprehensive Blocking for Intracellular Staining
Intracellular staining for stem cell markers presents unique challenges for blocking strategies. Permeabilization exposes a vastly expanded landscape of potential off-target epitopes, significantly increasing opportunities for non-specific binding [30] [33]. Additionally, the fixation process itself can alter protein structure and create new non-specific binding sites [33]. For these applications, researchers often benefit from an additional blocking step after permeabilization but before intracellular antibody incubation [30].
The choice of permeabilization method influences blocking strategy. When using saponin, which creates reversible pores in membranes, the permeabilizing agent must be maintained in all subsequent wash and antibody buffers to ensure continued access to intracellular targets [33] [34]. Stronger detergents like Triton X-100 or organic solvents like methanol create permanent permeability but may damage certain epitopes or denature protein-based fluorophores like PE and APC [33]. For transcription factors and nuclear markers common in stem cell research (e.g., NANOG), harsher permeabilization methods are often necessary to access nuclear targets [33].
Experimental Protocols
Basic Protocol: Surface Staining with Integrated Blocking
This optimized protocol provides a general-use approach for reducing non-specific interactions during surface staining in high-parameter flow cytometry [30].
Materials
- Mouse serum (e.g., Thermo Fisher, cat. no. 10410) [30]
- Rat serum (e.g., Thermo Fisher, cat. no. 10710C) [30]
- Tandem stabilizer (e.g., BioLegend, cat. no. 421802) [30]
- Brilliant Stain Buffer (e.g., Thermo Fisher, cat. no. 00-4409-75) or BD Horizon Brilliant Stain Buffer Plus (BD Biosciences, cat. no. 566385) [30]
- FACS buffer (PBS with 0.5-1% BSA or FCS, optionally with 0.1% sodium azide) [30] [11]
- Sterilin clear microtiter plates, 96-well V-bottom [30]
- Centrifuge and multichannel pipettes [30]
Procedure
- Prepare a blocking solution comprising rat serum, mouse serum, tandem stabilizer, and serum from any other host species represented in your antibody panel according to Table 3 [30].
- Dispense cells into V-bottom, 96-well plates (standardize cell numbers to reduce batch effects) [30].
- Centrifuge for 5 minutes at 300 × g, 4°C or room temperature, and remove supernatant [30].
- Resuspend cells in 20 µL blocking solution and incubate for 15 minutes at room temperature in the dark [30].
- While blocking, prepare surface staining master mix containing tandem stabilizer, Brilliant Stain Buffer (up to 30% v/v), and predetermined antibody concentrations in FACS buffer [30].
- Add 100 µL surface staining mix directly to each sample (without washing blocking solution) and mix by pipetting [30].
- Incubate for 1 hour at room temperature in the dark [30].
- Wash with 120 µL FACS buffer, centrifuge for 5 minutes at 300 × g, and discard supernatant [30].
- Repeat wash with 200 µL FACS buffer [30].
- Resuspend samples in FACS buffer containing tandem stabilizer at 1:1000 dilution and acquire on cytometer [30].
Table 3: Blocking Solution Formulation for Surface Staining
| Reagent | Dilution Factor | Volume (µL) for 1-mL Mix |
|---|---|---|
| Mouse serum | 3.3 | 300 |
| Rat serum | 3.3 | 300 |
| Tandem stabilizer | 1000 | 1 |
| Sodium azide (10%) | 100 | 10 (optional for short-term use) |
| FACS buffer | Remainder | 389 |
Advanced Protocol: Sequential Extracellular and Intracellular Staining
For simultaneous detection of cell surface markers and intracellular stem cell markers, this sequential protocol maximizes specificity for both compartments.
Materials
- Fixative: 1-4% paraformaldehyde (PFA) in PBS (ice-cold) [33]
- Permeabilization buffers: Methanol (ice-cold), Triton X-100 (0.1-0.3% in PBS/BSA), or saponin (0.1-0.5% in PBS/BSA) [33]
- Intracellular blocking buffer: 2-10% normal serum from antibody host species in permeabilization buffer [30] [11]
- Additional reagents from Basic Protocol
Procedure
- Complete surface staining protocol (steps 1-9 of Basic Protocol) including Fc blocking and surface antibody incubation [30] [33].
- Fix cells with 100 µL ice-cold 4% PFA for 20 minutes at room temperature [33].
- Wash with 200 µL PBS to remove excess fixative [33].
- Permeabilize cells using appropriate method:
- Methanol: Add 100 µL 90% ice-cold methanol, incubate 15 minutes on ice (not compatible with protein-based fluorophores) [33]
- Triton X-100: Add 100 µL 0.1-0.3% Triton X-100 in PBS/BSA, incubate 10 minutes at room temperature [33]
- Saponin: Add 100 µL 0.1% saponin in PBS/BSA, incubate 10 minutes at room temperature (include saponin in all subsequent buffers) [33]
- Centrifuge and resuspend cells in 20 µL intracellular blocking buffer [30].
- Incubate for 15 minutes at room temperature in the dark [30].
- Prepare intracellular staining mix with antibodies diluted in appropriate permeabilization buffer [30].
- Add 100 µL intracellular staining mix to each sample and incubate for 30-60 minutes at room temperature in the dark [30] [33].
- Wash twice with 200 µL permeabilization buffer [33].
- Wash once with 200 µL FACS buffer to remove permeabilization agents [33].
- Resuspend in FACS buffer with tandem stabilizer for acquisition [30].
Protocol for Stem Cell Marker Staining
This specialized protocol is adapted for the unique requirements of human induced pluripotent stem cells (iPSCs) and intracellular pluripotency markers [13].
Materials
- iPSC culture collected as single-cell suspension [13]
- EDTA solution for gentle cell detachment [13]
- Stem cell-specific viability dye (compatible with fixation)
- Pluripotency marker antibodies (surface and intracellular)
Procedure
- Culture iPSCs under standard conditions and harvest using gentle enzymatic or EDTA-based dissociation to preserve epitopes [13].
- Determine total cell number and check viability (aim for 90-95% viability) [11].
- Perform live/dead staining with a fixable viability dye according to manufacturer's protocol [11] [32].
- Block Fc receptors using species-appropriate blocking reagent for 15 minutes at room temperature [30] [13].
- Stain surface markers (e.g., CD9, CD24, CD133, SSEA-4) using Basic Protocol steps [13].
- Fix cells with 4% PFA for 20 minutes at room temperature [13] [33].
- Permeabilize with 0.1% Triton X-100 for 10 minutes at room temperature [33].
- Block with intracellular blocking buffer for 15 minutes at room temperature [30].
- Stain intracellular pluripotency markers (e.g., NANOG, OCT4, SOX2) for 30 minutes at room temperature [13].
- Wash twice with permeabilization buffer followed by one wash with FACS buffer [33].
- Resuspend in FACS buffer for acquisition on flow cytometer [13].
The Scientist's Toolkit: Essential Reagents
Table 4: Key Research Reagent Solutions for Blocking Strategies
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Fc Blocking Reagents | Normal sera (mouse, rat, human); Purified IgG; Anti-CD16/CD32 | Blocks Fc receptor binding | Match host species to primary antibodies; Use purified IgG for reduced variability [31] |
| Dye Stabilizers | Brilliant Stain Buffer; Tandem Stabilizer | Prevents dye-dye interactions and tandem degradation | Essential for Brilliant Violet dyes and tandem fluorophores; Include in storage buffer [30] |
| Fixation Reagents | 4% Paraformaldehyde (PFA); Methanol; Acetone | Preserves cellular structure and intracellular antigens | PFA preferred for surface epitopes; Methanol can unmask some phospho-epitopes [33] |
| Permeabilization Agents | Triton X-100; Saponin; Tween-20; Methanol | Enables antibody access to intracellular targets | Strong detergents for nuclear antigens; Mild detergents for cytoplasmic targets [33] |
| Viability Dyes | 7-AAD; DAPI; Fixable viability dyes | Distinguishes live from dead cells | Critical as dead cells bind antibodies non-specifically; Use fixable dyes for intracellular staining [11] [32] |
| Specialized Blockers | True-Stain Blocker; Oligo-Block; CellBlox | Blocks fluorophore-specific interactions | Address specific dye-cell interactions; Use when unusual staining patterns occur [31] |
Workflow Visualization
Effective blocking strategies are not merely optional refinements but essential components of rigorous flow cytometry panel design, particularly for challenging applications like intracellular stem cell marker detection. The integration of comprehensive Fc receptor blocking, fluorophore-specific stabilization, and targeted intracellular blocking significantly enhances data quality by reducing non-specific background while preserving specific signal. For researchers characterizing iPSCs and other stem cell populations, where accurate quantification of pluripotency markers directly impacts experimental conclusions, these protocols provide a validated framework for maximizing assay sensitivity and specificity. As flow cytometry continues to evolve toward higher parameter panels, the systematic implementation of these blocking strategies will remain fundamental to generating reproducible, publication-quality data in stem cell research and therapeutic development.
Within the evolving field of stem cell research, the precise identification and characterization of stem cells and their differentiated progeny are paramount. These rare cell populations, often residing in heterogeneous mixtures, are defined by the expression of specific intracellular transcription factors and proteins [10]. Flow cytometry stands as a powerful tool for this purpose, offering high-throughput, multi-parameter analysis at single-cell resolution [10]. However, the accurate detection of low-density intracellular markers presents a significant technical challenge. The success of such experiments hinges on a critical principle: the strategic pairing of dimly expressed targets with bright fluorophores to achieve a sufficient signal-to-noise ratio [35] [36]. This application note provides a detailed protocol and framework for researchers and drug development professionals to optimize fluorochrome selection for intracellular stem cell marker analysis, ensuring reliable and reproducible data.
The Critical Challenge of Intracellular Marker Detection
Stem cell research frequently involves the detection of intracellular proteins, such as transcription factors (e.g., Nanog, Oct-4) and other functional markers, which are often expressed at low levels [10] [35]. Unlike cell surface antigens, accessing these targets requires cells to be fixed and permeabilized, a process that can damage epitopes, increase autofluorescence, and alter light scatter properties [35]. Furthermore, the natural autofluorescence of cells, particularly at lower wavelengths, can mask the specific signal from a faint marker [37].
The antigen density is a primary consideration in panel design. Low-density antigens generate a weak fluorescence signal, making it difficult to distinguish positive cells from the negative population [35] [36]. To resolve this signal from background noise, it is essential to use the brightest possible fluorophores [36]. Using a dim fluorophore for a low-abundance marker will result in poor population resolution and potentially misleading data.
Principles of Fluorochrome Selection
Key Properties of Fluorochromes
When selecting fluorochromes, several properties must be evaluated to ensure optimal performance in intracellular staining applications:
- Brightness: The fluorescence intensity of a fluorochrome is paramount for detecting low-density markers. Brightness is a function of the fluorochrome's extinction coefficient and quantum yield [37].
- Photostability: Fluorochromes that are resistant to photobleaching maintain signal integrity over longer acquisition times, which is crucial for sorting or when analyzing rare populations [38].
- Spectral Overlap: Fluorochromes with broad emission spectra can spill over into adjacent detectors, necessitating compensation and potentially compromising data quality. Choosing fluorochromes with minimal spectral overlap simplifies panel design and improves resolution [36].
- Intracellular Performance: Some fluorochromes are more susceptible to the effects of fixation and permeabilization, which can quench their fluorescence or increase non-specific binding. It is essential to select dyes that have been validated for intracellular use [38].
Fluorochrome Classes and Their Applications
Fluorochromes can be broadly categorized into three classes, each with distinct characteristics [37] [38]:
- Traditional Organic Fluorophores: These include dyes like Fluorescein Isothiocyanate (FITC) and Phycoerythrin (PE). PE is exceptionally bright and is often recommended for low-density targets.
- Tandem Dyes: These are conjugates of two fluorophores (e.g., PE-Cy7, Brilliant Violet 785) where energy is transferred from a "donor" to an "acceptor," creating a new emission profile. While they expand the panel's options, tandem dyes can be sensitive to fixation and light exposure, potentially leading to decoupling and increased spillover [37] [38].
- Synthetic Dyes: Newer dyes, such as the BD Horizon RealBlue and RealYellow series, NovaFluor, and StarBright, are engineered for enhanced brightness, photostability, and lower spillover, making them excellent choices for complex intracellular panels [38].
Table 1: Performance Characteristics of Common Fluorochromes for Intracellular Staining
| Fluorochrome | Relative Brightness | Primary Laser(s) | Intracellular Performance | Photostability | Notes |
|---|---|---|---|---|---|
| PE | Very High [36] | Blue (488 nm) [36] | Good [38] | Moderate | Benchmark for brightness; ideal for rare antigens [36] |
| APC | High [36] | Red (633-640 nm) [36] | Good | Moderate | Excellent for low-density markers [36] |
| BD Horizon RealYellow 703 | High | Yellow-Green (561 nm) [38] | Excellent | High | Winner in photostability faceoff; minimal spillover change [38] |
| BD Horizon RealBlue 613 | High | Blue (488 nm) [38] | Excellent | High | Validated for intracellular staining of Granzyme B [38] |
| FITC | Medium [36] | Blue (488 nm) [36] | Good | Moderate | Susceptible to environmental pH [37] |
| PerCP | Low [36] | Blue (488 nm) [36] | Variable | Moderate | Avoid for critical low-density markers |
The following workflow diagram outlines the logical decision-making process for selecting the appropriate fluorochrome based on marker density and cellular localization:
Experimental Protocol: Intracellular Staining of Stem Cell Markers
This protocol provides a step-by-step methodology for the intracellular staining of transcription factors in human pluripotent stem cells, incorporating best practices for handling low-abundance targets.
Materials and Reagents
Table 2: Research Reagent Solutions for Intracellular Flow Cytometry
| Item | Function | Example Product/Catalog |
|---|---|---|
| Fixation Buffer | Crosslinks and preserves cellular proteins, halting metabolism and stabilizing the cell structure. | BD Phosflow Lyse/Fix Buffer (Cat. No. 558049) [38] |
| Permeabilization Buffer | Disrupts lipid membranes to allow intracellular antibody access while preserving light scatter properties. | BD Perm/Wash Buffer (Cat. No. 554723) [38] |
| Bright Fluorophore-Conjugated Antibodies | Specific detection of low-density intracellular antigens. | Antibodies conjugated to PE, APC, or RealYellow dyes targeting markers like Nanog, Oct-4 [10] [38] |
| Protein Transport Inhibitor | Blocks protein secretion for cytokine staining, allowing intracellular accumulation. | Brefeldin A [35] |
| Viability Dye | Distinguishes live from dead cells; critical for excluding false-positive signals from dead cells. | 7-AAD [36] or similar dye |
| Compensation Beads | Ultraviolet-compensation for multicolor panels; essential for calculating spillover between channels. | Antibody capture beads [36] |
Step-by-Step Procedure
Sample Preparation:
- Harvest stem cells using a gentle dissociation reagent to obtain a single-cell suspension. The requirement for a single-cell suspension is fundamental for accurate flow cytometry analysis [10].
- If analyzing induced cytokines, treat cells with the appropriate stimulus and add a protein transport inhibitor (e.g., Brefeldin A) for the final 4-6 hours of culture [35].
Viability Staining:
- Resuspend the cell pellet in a buffer containing a viability dye (e.g., 7-AAD) and incubate as per manufacturer's instructions. This step is performed prior to fixation to avoid non-specific staining of dead cells [36].
Fixation and Permeabilization:
- Fix cells by resuspending them in a commercially available fixation buffer (e.g., BD Phosflow Lyse/Fix Buffer) and incubating for 10-15 minutes at 37°C [38].
- Wash cells twice with a flow cytometry staining buffer.
- Permeabilize cells by resuspending the pellet in a permeabilization buffer (e.g., BD Perm/Wash Buffer). Incubate for 15-30 minutes on ice [38].
Intracellular Immunostaining:
- During permeabilization, prepare the antibody master mix in permeabilization buffer. For low-density markers, titrate the bright fluorophore-conjugated antibody beforehand to determine the optimal signal-to-noise ratio.
- Centrifuge the permeabilized cells and resuspend the pellet in the antibody mix.
- Incubate for 30-60 minutes in the dark at room temperature.
Wash and Acquisition:
- Wash cells twice with permeabilization buffer to remove unbound antibody.
- Resuspend the final cell pellet in a suitable acquisition buffer.
- Acquire data on a flow cytometer within a few hours. For experiments using tandem dyes, minimize light exposure to prevent degradation [38].
The workflow for the entire experimental procedure, from sample preparation to data acquisition, is summarized below:
Panel Design and Data Analysis
Optimizing Multicolor Panels
Designing a panel for intracellular stem cell markers requires careful strategic planning to account for spectral overlap and antigen density.
- Laser Compatibility: Ensure the fluorochromes selected are excited by the lasers available on your flow cytometer [36].
- Spectral Overlap Management: Use tools like online panel builders and spectral viewers to select fluorochromes with minimal emission spectrum overlap [35]. Avoid pairing a very bright fluorophore with a low-density marker if its spillover will interfere with the detection of another dim marker in a nearby channel [36].
- Density-Brightness Matching: Adhere to the core principle: assign the brightest fluorophores (e.g., PE, APC, RealYellow dyes) to the lowest density or most critical markers [38] [36]. For example, a key pluripotency transcription factor like Nanog should be detected with PE, while a highly expressed structural protein could be assigned to FITC.
Table 3: Example Panel for Human Pluripotent Stem Cell Characterization
| Target Marker | Marker Type & Density | Recommended Fluorochrome | Rationale |
|---|---|---|---|
| Nanog | Intracellular / Low | PE or BD Horizon RY703 | Maximum brightness for critical, low-abundance transcription factor [10] [38] |
| SSEA-4 | Surface / High | FITC or BV421 | High antigen abundance allows for a dimmer fluorophore [10] |
| TRA-1-60 | Surface / Medium | APC | Bright fluorophore for confident population identification [10] |
| Viability | N/A | Fixable Viability Dye eFluor 780 | Far-red dye to avoid interference with key markers |
Compensation and Controls
- Compensation Controls: For each fluorophore used, run a single-stained control. This control can be cells or compensation beads [36]. The positive signal must be at least as bright as in the experimental sample.
- Fluorophore-Matched Controls: The compensation control must use the same antibody-fluorophore conjugate as the experimental panel [36].
- Full Panel Validation: After setting compensation with controls, run an unstained control, a fluorescence-minus-one (FMO) control for each channel, and the fully stained sample to confirm proper panel performance and gating.
The accurate resolution of low-density intracellular markers is a cornerstone of advanced stem cell research. By understanding the properties of modern fluorochromes and adhering to a disciplined panel design strategy that matches bright fluorophores to challenging targets, researchers can unlock deeper insights into stem cell biology. The continued development of brighter, more stable dyes and sophisticated analytical instruments promises to further enhance our ability to characterize these rare and clinically vital cell populations with unprecedented precision.
The characterization of intracellular stem cell markers, such as transcription factors NANOG and other pluripotency factors, is fundamental to iPSC research, regenerative medicine, and drug development [13] [10]. Flow cytometry provides the high-throughput, single-cell resolution necessary for this task, but the data's validity hinges entirely on the implementation of appropriate experimental controls [32] [39]. For intracellular staining, where fixation and permeabilization steps increase background fluorescence and non-specific antibody binding, controls are not merely optional but essential [32]. They isolate specific signals from experimental noise, ensuring that the observed fluorescence accurately reflects the expression of the target intracellular antigen. This application note details the critical trio of controls—Unstained, Isotype, and Fluorescence Minus One (FMO)—within the context of a robust flow cytometry protocol for intracellular stem cell marker analysis, providing researchers with a framework for generating publication-quality data.
The Control Ecosystem in Flow Cytometry
In a multicolor flow cytometry experiment, different controls serve distinct and non-interchangeable purposes. They can be broadly categorized as instrumental controls, which ensure the cytometer is configured correctly, and experimental controls, which validate the staining specificity [39]. The following workflow illustrates how these critical controls are integrated into a typical experimental setup for analyzing intracellular stem cell markers.
Figure 1: Control Integration Workflow. This diagram outlines the sequential and parallel roles of critical controls in a flow cytometry experiment for intracellular staining. The process begins with experimental design and proceeds through the use of controls for instrument setup (Unstained, Single Fluorophore), gating strategy (FMO), and validation of staining specificity (Isotype), culminating in the analysis of the fully stained experimental sample.
Detailed Characterization of Critical Controls
Unstained Control
The unstained control consists of cells that have undergone the entire experimental procedure—including fixation and permeabilization for intracellular staining—but have not been incubated with any fluorescent antibodies [39]. This control is fundamental for identifying the innate autofluorescence of the cells, which can be substantial in fixed samples and can vary significantly between different stem cell lines [32] [39]. Its primary function is to establish the baseline fluorescence of the cells, which is used to set the photomultiplier tube (PMT) voltages for all fluorescent channels, ensuring the detector sensitivity is optimized to capture the true staining signal above the cellular background [39].
Isotype Control
An isotype control is an antibody that matches the experimental antibody in host species, immunoglobulin class and subclass, and fluorophore conjugation, but is raised against a target not present in the sample (e.g., a V5 tag in cells not engineered to express it) [39]. It is used to determine the level of non-specific antibody binding caused by interactions with cellular components, such as Fc receptors, or other hydrophobic or charged structures exposed during permeabilization [32]. It is critical to note that the isotype control should not be used to set the boundary between positive and negative populations (the "positive gate") [32]. Its proper use is to quantify and account for background fluorescence stemming from non-specific antibody binding, which can then be considered when interpreting the signal from the specific experimental antibody.
Fluorescence Minus One (FMO) Control
The FMO control is a sample stained with all antibodies in the multicolor panel except for one. For example, in a panel containing FITC, PE, and APC, the FMO control for the PE channel would contain FITC and APC, but not PE [32] [39]. This control is the gold standard for accurately setting positive gates in complex multicolor experiments. It accounts for the "fluorescence spread" or "spillover" from all other fluorophores in the panel into the channel of interest, which an unstained control cannot do [32]. Using an FMO control to establish gating thresholds prevents the misclassification of cells that appear dimly positive due to spectral overlap rather than true antigen expression.
Table 1: Summary of Critical Flow Cytometry Controls for Intracellular Staining
| Control Type | Purpose | Key Application | Limitations & Notes |
|---|---|---|---|
| Unstained [39] | Establish baseline autofluorescence and set PMT voltages. | Determine background from fixed/permeabilized cells. | Does not account for spectral overlap or non-specific antibody binding. |
| Isotype [32] [39] | Measure non-specific antibody binding (e.g., to Fc receptors). | Assess background from antibody-cell interactions. | Should not be used for setting positive gates; used to interpret specificity. |
| FMO [32] [39] | Accurately define positive/negative populations by accounting for spectral spillover. | Set correct gating boundaries in multicolor panels. | Resource-intensive; requires one sample per fluorophore in the panel. |
Comprehensive Protocol for Control Preparation in Intracellular Staining
This protocol is adapted from established methods for the analysis of undifferentiated stem cell markers [13] and general flow cytometry best practices [32] [39].
Basic Protocol 1: iPSC Culture and Preparation of Single-Cell Suspension
- Culture Human iPSCs: Maintain human induced pluripotent stem cells (iPSCs) under standard, feeder-free conditions using a defined matrix and mTeSR or equivalent medium. Ensure cells are in an undifferentiated state, as confirmed by morphology.
- Harvest Cells: Gently wash cells with DPBS without calcium and magnesium. Dissociate cells into a single-cell suspension using a gentle cell dissociation reagent (e.g., Accutase or EDTA). Avoid using trypsin if the intracellular epitope of interest is sensitive to its activity.
- Count and Aliquot: Count cells using an automated counter or hemocytometer. Aliquot a minimum of (0.5 - 1 \times 10^6) cells per control and experimental tube. Centrifuge at (300 \times g) for 5 minutes and aspirate the supernatant.
Basic Protocol 2: Fixation, Permeabilization, and Staining
- Fixation: Resuspend cell pellets in a freshly prepared, buffered 4% paraformaldehyde (PFA) solution. Incubate for 15-20 minutes at room temperature (protected from light).
- Washing: Add 2 mL of flow cytometry staining buffer (e.g., DPBS with 1% BSA and 0.1% sodium azide) to each tube. Centrifuge at (500 \times g) for 5 minutes and carefully decant the supernatant.
- Permeabilization: Resuspend cells thoroughly in 100 µL - 1 mL of a permeabilization buffer (e.g., 90% methanol, or a commercial saponin-based buffer). Incubate for 15-30 minutes on ice or at room temperature, as required by the intracellular target.
- Staining and Control Setup:
- Unstained Control: Resuspend one aliquot of cells in an appropriate volume (e.g., 100 µL) of staining buffer. Do not add any antibodies.
- Isotype Control: Resuspend one aliquot of cells in 100 µL of staining buffer containing the pre-titrated, fluorophore-conjugated isotype control antibody.
- FMO Controls: Prepare one tube for each fluorophore in your panel. For a given FMO control (e.g., FMO-PE), resuspend cells in a cocktail containing all antibodies except the PE-conjugated one.
- Fully Stained Experimental Sample: Resuspend cells in the complete antibody cocktail containing all fluorophore-conjugated antibodies against the intracellular targets (e.g., NANOG, SOX2, OCT4).
- Single Fluorophore Controls (for Compensation): For each fluorophore used in the panel, stain a separate aliquot of cells or compensation beads with only that single antibody [39].
- Incubation: Incubate all tubes for 30-60 minutes at room temperature in the dark.
- Final Wash: Add 2 mL of staining (and permeabilization) buffer to each tube, centrifuge, and aspirate the supernatant. Resuspend the final cell pellet in 200-300 µL of staining buffer for acquisition. Keep samples at 4°C and protected from light.
Basic Protocol 3: Flow Cytometry Acquisition and Data Analysis
- Instrument Setup: Start with the unstained control. Adjust FSC and SSC voltages to position the cell population on-scale. For each fluorescence detector, adjust the PMT voltage so that the autofluorescence signal for the unstained population is in the first decade of the logarithmic plot [39].
- Compensation: Using the single-color stained controls (cells or beads), run each sample and use the flow cytometer's software to calculate the compensation matrix. Apply this matrix to all samples, including the controls and experimental tubes [32].
- Gating Strategy:
- Use FSC-A vs. SSC-A to gate on single cells and exclude debris.
- Use the unstained control to confirm the autofluorescence baseline.
- Use the FMO control to set the positive gate for its corresponding channel. The boundary between negative and positive populations should be placed where the FMO histogram ends.
- Use the isotype control to contextualize the level of non-specific binding in the experimental sample.
- Analysis: Analyze the fully stained experimental sample using the gating strategy defined by the controls. Report the percentage and median fluorescence intensity (MFI) of positive cells for each intracellular marker.
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for Intracellular Flow Cytometry
| Item | Function/Description | Example Application |
|---|---|---|
| Validated Antibodies [32] | Antibodies specifically validated for specificity and lack of cross-reactivity in intracellular flow cytometry. | Critical for detecting intracellular transcription factors like NANOG in iPSCs [13]. |
| Fluorophore-Conjugated Antibodies [40] | Antibodies directly conjugated to fluorophores (e.g., Alexa Fluor 488, PE, APC). | Enable multiplexed detection of multiple intracellular markers simultaneously. |
| Fixation/Permeabilization Kits | Commercial kits providing optimized buffers for crosslinking and disrupting cell membranes. | Essential for allowing antibodies to access intracellular epitopes while preserving cell structure and light scatter properties. |
| Fc Receptor Blocking Reagent [32] | Reagent (e.g., purified human IgG) used to block Fc receptors on cells. | Reduces non-specific antibody binding, a critical step before intracellular staining. |
| Compensation Beads [32] [39] | Synthetic beads that bind antibodies, used to create single-color controls. | Allow for accurate compensation in multicolor panels when cell numbers are limited. |
| Cell Viability Dye [32] | Impermeable DNA-binding dye (e.g., 7-AAD, DRAQ7) to identify dead cells. | Dead cells exhibit high autofluorescence and non-specific binding; excluding them improves data quality. |
| Flow Cytometry Staining Buffer | Buffer (e.g., DPBS with BSA/serum) for antibody dilution and washing. | Preserves cell viability and health during staining procedures and reduces non-specific background. |
The rigorous application of Unstained, Isotype, and FMO controls is not a mere formality but the foundation of reliable flow cytometry data, especially in the challenging context of intracellular stem cell marker analysis. By systematically implementing the protocols and principles outlined in this application note, researchers can confidently distinguish true pluripotency marker expression from experimental artifacts, thereby generating robust, reproducible, and meaningful data to advance their research in stem cell biology and therapeutic development.
Solving Common Problems and Optimizing Signal-to-Noise in Intracellular Assays
In the field of flow cytometry for intracellular stem cell marker research, achieving high signal-to-noise ratio is paramount for accurate data interpretation. High background staining can obscure critical results, leading to false positives and compromised data integrity. This is particularly relevant when working with sensitive cell types like induced pluripotent stem cells (iPSCs), where precise characterization of undifferentiated markers defines their pluripotent status [13]. Two fundamental strategies to minimize background staining include Fc receptor (FcR) blocking to prevent non-specific antibody binding and rigorous antibody titration to determine optimal staining concentrations [41] [42]. This application note provides detailed protocols and methodologies to effectively implement these strategies, framed within the context of intracellular stem cell marker analysis.
Background and Principles
The Fc Receptor Interference Problem
Flow cytometry relies on the specific binding of fluorescently-labeled antibodies to cellular targets. However, the specificity of this binding is contingent upon the unique variable regions of each antibody clone. The constant Fc region of antibodies can non-specifically bind to a variety of immune cells expressing Fc receptors, including monocytes, macrophages, B lymphocytes, dendritic cells, and NK cells [41]. This non-specific binding generates increased background fluorescence and can create false positive populations in the analysis [41]. Although often associated with immune cells, Fc receptor-mediated binding is a critical consideration in stem cell research, particularly when analyzing heterogeneous populations or differentiated progeny that may express these receptors.
The Critical Role of Antibody Titration
Antibody titration is the systematic process of determining the reagent concentration that provides the best resolution between a positive signal and background noise [42]. Using an incorrect antibody concentration severely impacts data quality. Under-staining results in weak signals, high variability, and potential underestimation of cell populations expressing the target marker. Over-staining leads to non-specific binding, wasteful reagent use, and detector overloading, which increases spillover spreading in multicolor panels [42]. For stem cell research, where markers like NANOG, SOX2, and OCT4 require precise quantification to define pluripotency, optimal titration is not merely a recommendation but a necessity for reproducible and reliable results [13].
Research Reagent Solutions
The following table catalogues essential reagents required for effective Fc receptor blocking and antibody titration protocols.
Table 1: Key Research Reagents and Their Functions
| Reagent | Function/Application |
|---|---|
| Purified IgG | Blocks Fc receptors by saturating binding sites with inert immunoglobulin [41]. |
| Anti-Fc Receptor Antibodies | Specifically targets and blocks specific Fc receptor subtypes [41]. |
| Staining Buffer | Provides the ionic and protein base for antibody dilutions and cell washing [42]. |
| Monoclonal Antibodies | Specifically bind to unique epitopes on target antigens (e.g., stem cell markers) [42]. |
| V-bottom 96-well Plates | Ideal vessel for low-volume staining and serial dilution protocols [42]. |
| Fc Block (Commercial) | Ready-to-use solution containing antibodies or IgG for Fc receptor blockade [42]. |
Experimental Protocols
Protocol 1: Fc Receptor Blocking
This protocol outlines the procedure for blocking Fc receptors on cells prior to antibody staining.
- Cell Preparation: Harvest and wash the cells (e.g., iPSCs or differentiated progeny). Resuspend the cell pellet in cold staining buffer at a concentration of 2 × 10^6 cells/mL [42].
- Blocking Reagent Selection: Choose an appropriate blocking reagent:
- Option A (Common): Purified IgG (e.g., human, mouse, or rat) at a concentration of 0.5-1 µg per million cells [41].
- Option B (Specific): Species-specific anti-Fc receptor antibodies.
- Note: Fetal bovine serum (FBS), a common buffer component, has insufficient IgG content and is not an effective FcR blocker [41].
- Blocking Incubation: Add the selected blocking reagent directly to the cell suspension. Incubate for 10-15 minutes at 4°C. Crucially, do not wash the cells after this step. The blocker must remain in the solution during the subsequent antibody staining step to maintain receptor saturation [41].
- Proceed to Staining: Without washing, add the pre-titrated, fluorescently-labeled antibody cocktail directly to the blocked cell suspension and continue with the standard staining protocol.
Protocol 2: Antibody Titration
This detailed protocol describes an 8-point serial dilution to determine the optimal concentration for a flow cytometry antibody.
- Determine Stock Concentration: Consult the antibody's product sheet or Certificate of Analysis (CoA) to find the stock concentration (often in µg/µL or µg/mL) [42].
- Calculate Initial Dilution:
- For antibodies with concentration in mg/mL, a common starting point is 1000 ng/test in a final staining volume of 200 µL [42].
- Prepare the first dilution in a V-bottom 96-well plate.
- Perform Serial Dilutions:
- Add 150 µL of staining buffer to the remaining 7-11 wells.
- Using a multichannel pipette, mix the first well and transfer 150 µL to the second well. Mix thoroughly.
- Continue this 2-fold serial dilution across the plate, mixing between each transfer. Discard 150 µL from the final well [42].
- Cell Staining:
- Add 100 µL of cell suspension (containing 2 × 10^5 cells) to each antibody dilution well. The final volume will be 250 µL.
- Incubate for 20 minutes at room temperature in the dark.
- Centrifuge the plate at 400 × g for 5 minutes, decant the supernatant, and blot on a paper towel.
- Wash the cells twice by resuspending in 200 µL of staining buffer, centrifuging, and decanting [42].
- Resuspend the cells in a fixed volume of buffer for acquisition.
- Data Acquisition and Analysis:
- Acquire data on a flow cytometer, collecting a sufficient number of events for statistical robustness.
- For each dilution, analyze the median fluorescence intensity (MFI) of the positive population and the negative population.
- Calculate the Stain Index (SI) for each dilution: SI = (MFIpositive - MFInegative) / (2 × SD_negative) [42].
- Plot the Stain Index against the antibody concentration. The optimal titer is the point just before the SI plateaus, indicating saturation of binding sites without significant excess antibody [42].
Data Presentation and Analysis
Quantitative Titer Selection
The following table exemplifies the data generated from a hypothetical antibody titration experiment. The optimal concentration is identified by the peak Stain Index.
Table 2: Example Antibody Titration Data for an Intracellular Stem Cell Marker
| Antibody Concentration (ng/test) | MFI (Positive) | MFI (Negative) | Stain Index |
|---|---|---|---|
| 1000 | 8550 | 520 | 12.5 |
| 500 | 8100 | 480 | 13.8 |
| 250 | 7250 | 450 | 13.2 |
| 125 | 5800 | 420 | 10.7 |
| 62.5 | 3800 | 400 | 6.4 |
| 31.3 | 2100 | 390 | 3.2 |
| 15.6 | 950 | 380 | 1.4 |
| 7.8 | 550 | 375 | 0.4 |
In this example, 500 ng/test is the optimal concentration, as it yields the highest Stain Index, indicating the best separation between positive and negative signals.
Workflow and Pathway Diagrams
Diagram 1: Fc Block and Staining Workflow
Diagram 2: Impact of Antibody Concentration
In the analysis of intracellular stem cell markers, flow cytometry data can be compromised by weak or absent signals, leading to inaccurate assessment of pluripotency and differentiation status. This issue frequently originates from suboptimal permeabilization, which prevents antibody access to intracellular epitopes, and poor fluorochrome-antibody pairing, which fails to provide sufficient detection sensitivity. The integrity of high-quality induced pluripotent stem cells (iPSCs) relies on precise measurement of key markers; failure to optimize these parameters can obscure critical biological findings and compromise experimental reproducibility. This application note provides a systematic framework for troubleshooting and resolving signal deficiency in intracellular staining protocols, with specific application to stem cell research.
Analysis of Permeabilization Efficiency
Effective permeabilization creates sufficient pores in the cell membrane to allow antibody entry while preserving cellular structure and surface epitopes. Inadequate permeabilization is a primary cause of weak intracellular signal.
Permeabilization Methods and Their Impact
Different permeabilization agents work through distinct mechanisms, with varying effectiveness across cell types and target locations.
Table 1: Comparison of Permeabilization Methods for Intracellular Staining
| Method | Mechanism | Best For | Advantages | Limitations |
|---|---|---|---|---|
| Saponin | Cholesterol sequestration creating temporary pores | Cytoplasmic antigens, transcription factors | Gentle procedure; reversible pores preserve cell integrity | Inconsistent for nuclear targets; requires continuous presence in buffers |
| Triton X-100 | Solubilizes lipid membranes | Robust penetration for nuclear antigens | Strong, consistent permeabilization | Can damage surface epitopes; may alter light scatter properties |
| Tween-20 | Mild detergent action | Combined surface/intracellular staining | Compatible with transcriptomic preservation; gentle on epitopes | Weaker penetration for dense cellular compartments |
| Methanol | Lipid dissolution and protein precipitation | Nuclear antigens; cell cycle analysis | Excellent nuclear access; simultaneously fixes | Can cause hypotonic shock; alters light scatter properties |
A gentle yet effective approach combining 0.25% buffered paraformaldehyde fixation followed by 0.2% Tween-20 permeabilization has demonstrated excellent preservation of both intracellular antigen accessibility and cell surface epitopes, making it particularly suitable for simultaneous surface and intracellular staining of precious stem cell samples [43].
Systematic Troubleshooting for Permeabilization Issues
When facing weak intracellular signals, researchers should investigate these critical parameters:
Permeabilization Agent Concentration and Timing: Optimize both concentration and incubation duration. While stronger detergents (e.g., Triton X-100) provide more robust penetration, they may compromise surface epitopes and increase background. For delicate stem cell markers, gentler methods (Tween-20, saponin) often yield superior results with proper optimization [44].
Fixation Compatibility: Ensure fixative choice aligns with permeabilization method. Aldehyde-based fixatives (paraformaldehyde) preserve epitopes better for subsequent staining than organic solvents (methanol). However, methanol-free formaldehyde is recommended to prevent premature permeabilization before sufficient cross-linking occurs [44].
Cellular Compartment Considerations: Nuclear targets (e.g., transcription factors) frequently require stronger permeabilization (methanol, Triton X-100) compared to cytoplasmic antigens. For large transcription factor complexes or protein aggregates, consider combining detergents with enzymatic methods [45].
Validation of Permeabilization Efficiency: Include positive controls with abundantly expressed intracellular proteins (e.g., structural proteins) to verify successful permeabilization independent of target antigen expression levels.
Strategic Fluorochrome Selection and Optimization
Matching fluorochrome brightness to antigen abundance is critical for detecting low-expression intracellular stem cell markers, particularly in highly autofluorescent cell types.
Fluorochrome Brightness and Antigen Matching
The fundamental principle for intracellular staining is pairing the brightest fluorochromes with the most weakly expressed targets [44]. This approach compensates for both low antigen density and signal reduction that may occur during permeabilization.
Table 2: Fluorochrome Selection Guide for Intracellular Stem Cell Markers
| Fluorochrome | Relative Brightness | Recommended Application | Compatibility with Permeabilization | Special Considerations |
|---|---|---|---|---|
| PE & PE Tandems | Very bright | Low-abundance transcription factors (e.g., NANOG, SOX2) | High (with saponin/Tween-20) | Large size may hinder nuclear access; prone to degradation [30] |
| APC & APC Tandems | Bright | Moderate to low abundance targets | High | More stable than PE tandems; better for long experiments |
| FITC | Moderate | High abundance structural proteins | High | Susceptible to cellular autofluorescence |
| Brilliant Violet dyes | Bright to very bright | Low abundance targets in autofluorescent cells | Variable | Check polymer dye compatibility with permeabilization methods [30] |
| Alexa Fluor dyes | Bright | General intracellular applications | High | Superior photostability; various brightness options |
For detecting crucial pluripotency markers like NANOG in iPSCs, which typically exhibit low expression levels, the brightest fluorochromes (PE, APC, or Brilliant Violet conjugates) are essential for clear resolution above background [13].
Addressing Fluorochrome-Related Challenges
Intracellular staining presents unique challenges for fluorochromes that must be addressed during experimental design:
Tandem Dye Stability: Tandem fluorochromes (e.g., PE-Cy7) are particularly susceptible to degradation, especially under suboptimal storage conditions or in the presence of certain fixation methods. Always use tandem dyes from the same manufacturing lot for all experiments and compensation controls [46]. Include tandem stabilizer in staining buffers to prevent dissociation [30].
Spectral Interactions: Dye-dye interactions can occur with certain fluorochrome families (Brilliant Violet, NovaFluors), potentially causing artificial signal correlation or suppression. Use specific staining buffers (Brilliant Stain Buffer) designed to minimize these interactions in multiplexed panels [30].
Size Considerations: Large fluorochrome-antibody conjugates (particularly tandems and polymer dyes) may have limited access to some intracellular compartments, especially in densely packed nuclear regions. For such targets, consider smaller fluorochromes (e.g., Alexa Fluor 488, FITC) despite their lower brightness [44].
Integrated Experimental Protocols
Simultaneous Surface and Intracellular Staining Protocol
This optimized protocol enables efficient detection of both surface and intracellular stem cell markers while minimizing cell loss, adapted for iPSC analysis [13] [14].
Materials:
- Fc receptor blocking solution (commercial Fc block or 10% serum from antibody host species)
- Fixation buffer (0.25-4% paraformaldehyde in PBS)
- Permeabilization buffer (0.2% Tween-20 in PBS or commercial perm buffer)
- Staining buffer (PBS with 1% BSA and 0.1% sodium azide)
- Fluorochrome-conjugated antibodies against surface and intracellular targets
- Viability dye (fixable viability dye for live/dead discrimination)
Procedure:
- Cell Preparation: Harvest iPSCs using gentle enzyme-free dissociation methods to preserve surface epitopes. Count and adjust concentration to 1-5×10^6 cells/mL [13].
- Fc Receptor Blocking: Resuspend cell pellet in 20μL blocking solution. Incubate 15 minutes at room temperature in the dark [30].
- Surface Staining (Optional): For surface markers only, proceed with antibody incubation for 30-60 minutes at 4°C, wash, then fix with 0.25% PFA for 1 hour at 4°C [43].
- Simultaneous Fixation and Permeabilization: For combined surface/intracellular staining, fix cells with 0.25% PFA for 1 hour at 4°C [43]. Permeabilize with 0.2% Tween-20 for 15 minutes at 37°C [43].
- Antibody Staining: Add premixed antibody cocktail against both surface and intracellular targets diluted in permeabilization buffer. Incubate 30-60 minutes at 4°C in the dark.
- Washing and Analysis: Wash twice with permeabilization buffer, then once with staining buffer. Resuspend in staining buffer with tandem stabilizer if using tandem dyes [30]. Acquire immediately on flow cytometer.
This simultaneous staining approach demonstrates comparable performance to traditional sequential methods while reducing cell loss by approximately 7-10%—a significant advantage when working with rare stem cell populations [14].
Permeabilization Optimization Workflow
Fluorochrome Selection Algorithm
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Intracellular Staining Optimization
| Reagent Category | Specific Products | Function | Application Notes |
|---|---|---|---|
| Fc Blocking Reagents | Normal serum from antibody host species; Commercial Fc receptor blocks | Reduces non-specific antibody binding via Fc receptors | Critical for hematopoietic cells; use serum matching antibody species [30] [47] |
| Fixation Agents | Paraformaldehyde (0.25-4%); Methanol-free formaldehyde | Preserves cellular structure and antigen integrity | Aldehyde fixatives better for surface epitopes; methanol-free recommended [44] |
| Permeabilization Detergents | Saponin (0.1-0.5%); Tween-20 (0.05-0.2%); Triton X-100 (0.1-0.5%) | Creates membrane pores for antibody access | Saponin for gentle cytoplasmic staining; Triton for nuclear targets [43] [44] |
| Tandem Stabilizers | Commercial tandem dye stabilizers | Prevents degradation of tandem fluorochromes | Essential for PE-Cy5, PE-Cy7, APC-Cy7 conjugates [30] |
| Brilliant Stain Buffers | BD Horizon Brilliant Stain Buffer; Plus variant | Prevents polymer dye interactions | Required for panels containing Brilliant Violet dyes [30] |
| Viability Dyes | Fixable viability dyes (e.g., eFluor, Zombie dyes) | Identifies and excludes dead cells | Reduces non-specific binding; choose fixable dyes for intracellular work [47] [44] |
Optimizing permeabilization and fluorochrome selection represents a critical pathway to resolving weak signal issues in intracellular stem cell marker analysis. Systematic evaluation of permeabilization methods based on target localization, combined with strategic pairing of antigen abundance with fluorochrome brightness, enables robust detection of even low-expression transcription factors essential for pluripotency assessment. The integrated protocols and troubleshooting frameworks presented here provide researchers with a comprehensive approach to enhancing signal detection, ultimately supporting more accurate characterization of stem cell populations and their differentiation states. As flow cytometry continues to advance toward higher parameter panels, these fundamental optimization principles remain essential for generating high-quality, reproducible data in stem cell research and drug development applications.
In the realm of flow cytometry, particularly within the sophisticated context of intracellular stem cell marker research, the exclusion of dead cells is not merely a recommendation but an essential component of experimental integrity. Dead cells compromise data quality through non-specific antibody binding and increased autofluorescence, ultimately reducing the dynamic range and obscuring the detection of weakly positive markers crucial for characterizing pluripotent status [48] [49]. For researchers investigating the expression of undifferentiated stem cell markers in induced pluripotent stem cells (iPSCs), this fidelity is paramount, as the accurate verification of pluripotency hinges on high-quality, reproducible data [13]. The incorporation of viability dyes provides a robust mechanism to identify and electronically exclude these compromised cells during analysis, thereby ensuring that subsequent gating strategies and data interpretation reflect true biological signals rather than technical artifacts. This application note details the strategic selection and implementation of viability dyes within a flow cytometry workflow designed for intracellular stem cell marker analysis, providing researchers with validated protocols to enhance the reliability of their findings.
Classes of Viability Dyes and Their Mechanisms of Action
Viability dyes function by exploiting the fundamental physiological difference between live and dead cells: the integrity of the cell membrane. Live cells possess intact membranes that selectively exclude certain compounds, whereas dead cells have compromised membranes that permit free dye entry. The choice of dye is primarily determined by the experimental requirements, most notably whether the protocol involves fixation and permeabilization for intracellular staining. The two principal classes of dyes are DNA-binding dyes and amine-reactive fixable viability dyes.
DNA-binding dyes, such as Propidium Iodide (PI) and 7-Aminoactinomycin D (7-AAD), are impermeant to live cells but readily enter dead cells, intercalating into double-stranded nucleic acids and producing a strong fluorescent signal [48] [49]. While effective for simple live/dead discrimination in surface-staining-only assays, a significant limitation is their incompatibility with intracellular staining protocols. The fixation and permeabilization steps required to access intracellular targets render all cells permeable, allowing these dyes to stain every cell and obliterate the distinction between live and dead populations [48] [11].
For intracellular staining protocols, amine-reactive fixable viability dyes (FVDs) are the definitive solution. These dyes covalently bind to amine groups on both extracellular and intracellular proteins. In a live cell, the dye only accesses surface amines, resulting in a low level of staining. In a dead cell, with its compromised membrane, the dye penetrates and labels the abundant intracellular amines, resulting in bright staining [48] [49]. This differential staining is permanently fixed upon cross-linking with aldehydes, allowing the dead cell population to be identified even after the subsequent permeabilization steps required for staining intracellular markers like NANOG or other transcription factors [48] [13] [11].
Table 1: Comparison of Major Viability Dye Classes
| Feature | DNA-Binding Dyes (PI, 7-AAD) | Fixable Viability Dyes (FVDs) |
|---|---|---|
| Mechanism of Action | Intercalation into double-stranded DNA/RNA [48] | Covalent binding to cellular amines (proteins) [48] [49] |
| Compatibility with Fixation/Permeabilization | No | Yes |
| Compatibility with Intracellular Staining | No | Yes |
| Typical Staining Time | 5-15 minutes [48] | ~30 minutes [48] |
| Key Advantage | Simple, fast protocol; low cost | Essential for any protocol involving cell fixation |
| Primary Disadvantage | Useless after fixation | Higher cost; requires proper storage (-70°C) [48] |
Experimental Protocols for Viability Staining
The following protocols have been adapted and optimized from best practices for flow cytometry, with special consideration for applications in stem cell research [48] [13] [11].
Protocol A: Staining with DNA-Binding Dyes (e.g., PI or 7-AAD)
This protocol is suitable only for experiments involving cell surface staining without fixation.
- Sample Preparation: After staining cells for surface antigens, wash the cells 1-2 times with a flow cytometry staining buffer by centrifuging at 200-600 × g for 5 minutes [48] [11].
- Dye Addition: Resuspend the cell pellet in an appropriate volume of buffer. Add 5 µL of Propidium Iodide or 7-AAD Staining Solution per 100 µL of cell suspension [48].
- Incubation: Incubate the cells for 5–15 minutes on ice or at room temperature. Protect the samples from light.
- Critical Note: Do not wash the cells after adding the dye. The dye must remain in the buffer during data acquisition to maintain dead cell labeling [48].
- Acquisition: Analyze the samples by flow cytometry immediately. Cells should be analyzed within 4 hours due to adverse effects on cell viability from prolonged dye exposure [48].
Protocol B: Staining with Fixable Viability Dyes (FVDs)
This is the required protocol for any experiment involving intracellular staining, such as for undifferentiated stem cell markers.
- Cell Preparation and Washing: Harvest and prepare a single-cell suspension of iPSCs. Wash the cells twice in azide-free and protein-free PBS to remove any exogenous amines that would quench the dye reaction [48].
- Dye Staining: Resuspend the cell pellet at a concentration of 1–10 × 10^6 cells/mL in azide-free, protein-free PBS. Add 1 µL of Fixable Viability Dye stock solution per 1 mL of cells and vortex immediately to ensure rapid and uniform mixing [48].
- Incubation: Incubate the cell-dye mixture for 30 minutes at 2–8°C. Protect from light throughout the incubation.
- Washing: Wash the cells 1-2 times with a protein-containing flow cytometry staining buffer to remove any unreacted dye [48].
- Continuation of Staining: The cells are now ready for subsequent steps. Proceed with surface antibody staining, followed by fixation and permeabilization for intracellular antibody staining, according to your established protocol [13] [11].
The Scientist's Toolkit: Essential Reagents and Materials
The following table catalogs the key reagents required for successfully incorporating viability dyes into a flow cytometry panel for stem cell research.
Table 2: Research Reagent Solutions for Viability Staining
| Reagent / Material | Function / Purpose | Example Products / Notes |
|---|---|---|
| Fixable Viability Dyes (FVDs) | Irreversibly labels dead cells for exclusion in fixed/permeabilized samples. | eFluor 450, 506, 780; Zombie dyes; Ghost dyes [48] [49]. |
| DNA-Binding Dyes (PI/7-AAD) | Labels dead cells in live-cell, surface-staining-only assays. | Propidium Iodide (cat. no. 00-6990), 7-AAD (cat. no. 00-6993) [48]. |
| Flow Cytometry Staining Buffer | Washing and suspension buffer; protein content can quench FVDs if present during staining. | Use amine-free PBS for FVD staining step; protein-based buffer for washes after [48]. |
| FcR Blocking Reagent | Prevents non-specific antibody binding to Fc receptors, improving signal-to-noise. | Human IgG, mouse anti-CD16/CD32, or 2-10% goat serum [11]. |
| Fixation & Permeabilization Kit | Preserves cell structure and allows antibody access to intracellular targets. | Commercial kits (e.g., ab185917) ensure optimal results for nuclear targets like NANOG [11]. |
Panel Design and Practical Considerations for Stem Cell Research
Integrating a viability dye into a multi-color flow cytometry panel requires strategic planning to ensure optimal data quality. The core principle is to select a viability dye whose emission spectrum does not overlap with the fluorochromes used to detect key biological markers [11]. For the characterization of undifferentiated stem cell markers, which often includes surface antigens (e.g., TRA-1-60, SSEA-4) and critical intracellular transcription factors (e.g., NANOG, OCT4), the bright signal from the viability dye should be placed in a channel that does not conflict with these detectors [13] [50].
Furthermore, proper compensation is critical in multi-color panels. The broad emission spectra of dyes like PI can cause significant spillover into adjacent channels. It is recommended to use a sample of the cells of interest, stained with the viability dye only, for setting compensation controls [48] [51]. When working with iPSC cultures where the baseline dead cell population may be low (<5%), a controlled compensation control can be created by heat-killing a small aliquot of cells (e.g., 65°C for 1 minute) and mixing them 1:1 with live cells before staining with the FVD [48]. This ensures a robust positive population for accurate compensation settings.
The systematic incorporation of viability dyes is a non-negotiable aspect of a rigorous flow cytometry protocol, especially in sensitive applications like the immunophenotyping of undifferentiated stem cells. By understanding the distinct properties and applications of DNA-binding versus fixable viability dyes, and by adhering to the optimized protocols detailed herein, researchers can effectively mitigate the confounding effects of dead cells. This practice significantly enhances data quality, improves the reliability of pluripotency status assessments, and ensures that conclusions drawn from complex intracellular staining experiments are built upon a foundation of robust, high-fidelity data.
Preventing Tandem Dye Degradation and Managing Dye-Dye Interactions
In high-parameter flow cytometry, particularly for intracellular stem cell marker research, the integrity of fluorescent signals is paramount for accurate data interpretation. Two significant technical challenges that compromise data quality are tandem dye degradation and dye-dye interactions. Tandem dyes, consisting of a donor fluorophore coupled to an acceptor molecule, are susceptible to breakdown, leading to erroneous signal detection [30]. Simultaneously, certain dye classes, especially polymer-based fluorophores, engage in intermolecular interactions that create spectral artifacts independent of biological binding [52]. For researchers working with precious samples like induced pluripotent stem cells (iPSCs), understanding and mitigating these phenomena is crucial for reliable detection of undifferentiated stem cell markers such as NANOG, as variations can profoundly impact experimental conclusions [13].
This application note provides detailed methodologies to prevent tandem dye degradation and manage dye-dye interactions within the context of intracellular staining protocols for stem cell research. By implementing these optimized procedures, researchers can significantly improve signal-to-noise ratios, enhance measurement sensitivity, and generate more reproducible data for drug development applications.
Mechanisms and Impacts of Dye-Related Artifacts
Tandem Dye Degradation
Tandem fluorophores function through Fluorescence Resonance Energy Transfer (FRET), where a donor fluorophore (e.g., PE or APC) transfers energy to an acceptor molecule (e.g., Cy7 or Cy5.5). Chemical or physical stress can break the covalent bond linking these components, causing the donor to emit at its native wavelength rather than the intended tandem emission spectrum [30]. This breakdown results in false-positive signals in channels detecting the donor fluorophore, leading to misidentification of cell populations. The degradation is influenced by multiple factors:
- Fixation Conditions: Extended exposure to formaldehyde-based fixatives accelerates tandem breakdown.
- Light Exposure: Photobleaching dissociates tandem complexes, particularly during prolonged staining incubations.
- Cellular Environment: Breakdown rates are higher on monocytes compared to lymphocytes, suggesting cell-type specific interactions [53].
Dye-Dye Interactions
Dye-dye interactions represent a distinct challenge where certain fluorophores interact directly, independent of their antibody conjugates. This phenomenon is particularly prominent with Brilliant Violet and Super Brilliant polymer dyes, which can form aggregates through hydrophobic and electrostatic interactions [52]. These interactions create false correlations between markers, potentially suggesting biological co-expression where none exists. The mechanisms differ from spectral overlap, as they occur prior to instrumental detection and cannot be corrected through compensation algorithms [30].
Table 1: Common Dye-Related Artifacts and Their Consequences
| Artifact Type | Primary Dyes Affected | Manifestation | Impact on Data |
|---|---|---|---|
| Tandem Degradation | PE-Cy7, APC-Cy7, PE-Cy5, PerCP-Cy5.5 | False signal in donor channel | Population misidentification, increased background |
| Cyanine Dye Binding | PE-Cy5, PE-Cy7, APC-Cy7 | Non-specific monocyte/macrophage binding | False-positive staining on specific cell types |
| Polymer Dye Interaction | Brilliant Violet dyes, Super Bright dyes | Correlated signals between markers | Artificial co-expression patterns |
The following diagram illustrates the mechanism of tandem dye degradation and the resulting detection artifacts:
Research Reagent Solutions
A strategic combination of specialized blocking buffers and staining reagents is essential for mitigating dye-related artifacts. The table below outlines key solutions validated for high-parameter flow cytometry applications:
Table 2: Essential Reagents for Managing Dye Artifacts
| Reagent | Composition | Primary Function | Application Specifics |
|---|---|---|---|
| Tandem Stabilizer | Proprietary stabilizing compounds | Prevents chemical dissociation of tandem dyes | Add to staining buffer (1:1000) and sample resuspension buffer [30] |
| Brilliant Stain Buffer | Polyethylene glycol (PEG) and proprietary agents | Disrupts polymer dye-dye interactions | Use at 30% (v/v) in staining mix; titrate to 1/4 concentration for cost savings [53] |
| Fc Receptor Blocking Solution | Normal serum (species-matched to antibodies) | Blocks non-specific antibody binding via Fc receptors | Prepare with 300μl mouse serum + 300μl rat serum per 1ml buffer [30] |
| CellBlox | Proprietary blocking molecules | Specifically reduces monocyte/macrophage binding of cyanine dyes | Essential for NovaFluor dyes; use per manufacturer instructions [30] |
| Species-Matched Sera | Normal serum from antibody host species | Reduces non-specific antibody binding | Use at 1:3.3 dilution in blocking solution [30] |
Optimized Experimental Protocols
Surface Staining Protocol with Enhanced Blocking
This protocol provides an optimized approach for surface staining of stem cell markers while minimizing dye-related artifacts:
Sample Preparation:
- Dispense cells into V-bottom 96-well plates (1×10⁶ cells/well)
- Centrifuge at 300 × g for 5 minutes at 4°C and remove supernatant [30]
Blocking Step:
- Resuspend cells in 20μl blocking solution (Table 2)
- Incubate 15 minutes at room temperature in the dark [30]
Staining Master Mix Preparation:
- Prepare surface staining mix containing:
- 30% Brilliant Stain Buffer (v/v)
- Tandem stabilizer (1:1000 dilution)
- Titrated antibody cocktails
- FACS buffer to volume [30]
- Prepare surface staining mix containing:
Staining Procedure:
- Add 100μl staining mix to each sample
- Incubate 1 hour at room temperature in the dark
- Wash with 120μl FACS buffer, centrifuge 5 minutes at 300 × g
- Repeat wash with 200μl FACS buffer [30]
Sample Resuspension:
- Resuspend in FACS buffer containing tandem stabilizer (1:1000)
- Acquire immediately or fix for short-term storage [30]
Simultaneous Intracellular and Surface Staining for Stem Cell Markers
For detecting intracellular stem cell markers (e.g., NANOG) alongside surface antigens:
Fixation:
- Fix cells immediately after surface staining using 4% methanol-free formaldehyde
- Incubate 15 minutes at room temperature [54]
Permeabilization and Simultaneous Staining:
- Permeabilize cells using ice-cold 90% methanol (add drop-wise while vortexing)
- Incubate 30 minutes on ice
- Prepare intracellular staining mix containing:
- Permeabilization buffer
- Intracellular antibodies (anti-NANOG, etc.)
- Tandem stabilizer (1:1000)
- Stain simultaneously for surface and intracellular markers for 1 hour at room temperature [14]
Validation:
- Fixed unfrozen samples show comparable performance to fresh samples with only 7-10% reduction in cell recovery [14]
The following workflow diagram outlines the optimized protocol for simultaneous intracellular and surface staining:
Panel Design and Technical Controls
Strategic Fluorophore Selection and Placement
Panel design decisions significantly impact susceptibility to dye artifacts:
- Assign Tandem Dyes to Highly Expressed Markers: This ensures sufficient signal intensity despite potential degradation [53]
- Position Tandem Dyes Post-Fixation: When possible, apply tandem-conjugated antibodies after fixation to reduce breakdown [53]
- Avoid Incompatible Dye Combinations: Separate Brilliant polymer dyes in panel design to minimize interactions [52]
- Match Fluorophore Brightness to Antigen Density: Use bright fluorophores (PE, APC) for low-density targets and dimmer fluorophores (FITC) for highly expressed markers [54]
Essential Experimental Controls
Implement rigorous controls to identify and account for residual dye artifacts:
- Fluorescence Minus One (FMO) Controls: Critical for establishing boundaries between positive and negative populations, especially for low-abundance stem cell markers [55]
- Single-Stain Controls: Prepare using the same cell type and processing as experimental samples [55]
- Unstained Controls: Account for cellular autofluorescence, which varies by cell type and treatment [55]
- Isotype Controls: Matched to species, immunoglobulin class, and fluorophore conjugation [55]
Table 3: Troubleshooting Guide for Dye-Related Issues
| Problem | Possible Cause | Solution |
|---|---|---|
| High background in donor channel | Tandem dye degradation | Add fresh tandem stabilizer; reduce light exposure; shorten fixation time |
| Correlated signals between markers | Dye-dye interactions | Increase Brilliant Stain Buffer concentration; redesign panel to separate problematic dyes |
| Non-specific monocyte staining | Cyanine dye binding | Incorporate CellBlox or similar monocyte blocking reagent |
| Poor signal-to-noise ratio | Insufficient Fc blocking | Increase concentration of species-matched serum; extend blocking incubation |
| Loss of resolution in highly multiplexed panels | Multiple interacting artifacts | Implement strategic panel splitting; use FMx controls to identify interactions |
Successful management of tandem dye degradation and dye-dye interactions requires a comprehensive approach spanning reagent selection, protocol optimization, and strategic panel design. For intracellular stem cell marker research, where population purity and characterization accuracy are paramount, implementing these detailed protocols significantly enhances data reliability. The combination of chemical stabilizers, strategic blocking reagents, optimized staining methodologies, and appropriate controls provides researchers with a robust framework for generating high-quality flow cytometry data essential for drug development and regenerative medicine applications.
Managing Suboptimal Scatter Properties and Instrument Clogging
In the field of intracellular stem cell marker research, the quality of flow cytometry data is paramount. Suboptimal light scatter properties and frequent instrument clogging represent two significant technical challenges that can compromise data integrity, leading to inaccurate immunophenotyping and erroneous conclusions. These issues are particularly prevalent when working with complex samples such as primary neural stem cells or densely cultured hematopoietic precursors, where cellular heterogeneity and debris are common [56]. Scatter profiles, which provide crucial information about cell size (forward scatter, FSC) and granularity/complexity (side scatter, SSC), serve as the primary gating reference for identifying viable cell populations of interest. When these profiles are poorly defined due to sample preparation artifacts or instrument malfunction, the entire analytical workflow is undermined. This application note details standardized protocols to identify, troubleshoot, and prevent these issues, ensuring the generation of high-quality, reproducible data for critical drug development and research applications.
Core Challenges and Underlying Causes
The Impact of Suboptimal Scatter Properties
Light scatter properties form the foundational step of flow cytometry analysis, enabling researchers to distinguish between different cell types and exclude debris prior to fluorescence analysis. Suboptimal scatter profiles manifest as poorly defined cell populations on FSC vs. SSC plots, excessive debris, and inconsistent population boundaries between samples. In the context of stem cell research, where target populations may be rare and heterogeneous, this lack of clarity can lead to the inadvertent exclusion of target cells or inclusion of non-viable cells and debris [56] [57]. The subsequent fluorescence analysis is thereby compromised, as gating strategies become unreliable and non-reproducible.
Several factors contribute to poor scatter resolution, including:
- Cell Clumping: Inadequate tissue disaggregation or improper pipetting during sample preparation creates cell aggregates that obstruct the flow cell and produce aberrant scatter signals [58].
- Non-Viable Cells: Dead and dying cells exhibit altered light scattering compared to healthy cells, broadening population distributions and increasing background noise [11].
- Sample Processing Artifacts: Over-digestion with enzymes like trypsin during harvesting from culture vessels can damage surface epitopes and alter cellular morphology, thereby changing scatter characteristics [56]. Excessive centrifugation force can also lyse cells, creating debris.
- Fixation and Permeabilization Effects: When performing intracellular staining for stem cell markers, the required fixation and permeabilization steps significantly affect light scatter profiles. For example, methanol-based fixatives can increase cell granularity, altering SSC properties [11]. Researchers must re-establish gating boundaries after these processing steps.
Instrument Clogging: Causes and Consequences
Instrument clogging occurs when particulate matter in the sample suspension obstructs the narrow fluidics path of the flow cytometer, most commonly at the sample injection port (SIP). This disruption in laminar flow produces erratic fluidics, leading to inconsistent sample core stream, unreliable measurements, and potentially complete cessation of data acquisition [59]. In severe cases, clogs require intensive maintenance procedures that result in significant instrument downtime, delaying critical experiments.
The primary causes of clogging include:
- Inadequate Sample Filtration: Failure to filter cell suspensions through an appropriate mesh (typically 40-70μm) before injection allows cell clumps and large debris to enter the fluidics system [58] [60].
- High Cell Concentration: Samples exceeding recommended concentrations (typically >1×10^7 cells/mL) increase the likelihood of coincident events and aggregation within the tubing [11].
- Sticky Samples: Certain cell types, particularly those undergoing apoptosis or containing sticky intracellular components released during permeabilization, are more prone to adhere to fluidics components [60]. Residual fibrin in poorly processed blood samples can also cause clogs.
- Crystalline Precipitates: Precipitation of salts or fixatives (e.g., paraformaldehyde) in storage buffers can introduce particulate matter that obstructs fluidics.
Comprehensive Experimental Protocols
Protocol for Optimizing Sample Scatter Properties
This protocol is designed for harvesting and preparing neural stem cell cultures to preserve native scatter characteristics and minimize debris, based on established methodologies for challenging cell types [56].
Materials Required:
- Fresh Cell Culture: Neural stem cells or other relevant stem cell population
- Mg²⁺/Ca²⁺-free Phosphate Buffered Saline (PBS)
- Appropriate detachment reagent: Trypsin replacement or Accutase
- Flow Buffer: PBS with 2% fetal bovine serum (FBS)
- Cell Strainer: 40μm nylon mesh
- Viability Dye: 7-AAD, propidium iodide, or similar DNA-binding dye [58]
- Centrifuge and refrigerated swinging-bucket rotor
- Polystyrene round-bottom tubes (12×75mm)
Procedure:
- Microscopic Assessment: Prior to harvesting, examine the culture using phase-contrast microscopy to assess confluence and visually identify contamination or excessive cell death [56].
- Gentle Cell Harvesting:
- Gently wash the adherent culture twice with room-temperature Mg²⁺/Ca²⁺-free PBS to remove residual media and detached cells [56].
- Add pre-warmed (37°C) trypsin replacement or Accutase, using sufficient volume to cover the monolayer.
- Incubate at 37°C for the minimum time required for detachment (typically 2-5 minutes), gently tapping the vessel to aid dispersal. Avoid over-digestion, which promotes cell surface damage and clumping.
- Quench the enzymatic reaction by adding twice the volume of ice-cold Flow Buffer (2% FBS in PBS).
- Single-Cell Suspension Preparation:
- Gently pipette the cell suspension to mechanically disaggregate clusters, avoiding vortexing or vigorous shaking that could damage cells [58].
- Pass the suspension through a 40μm cell strainer into a clean tube to remove remaining aggregates and debris [58] [60].
- Centrifuge at 200×g for 5 minutes at 4°C to pellet cells. Higher g-forces may lyse cells and create debris.
- Viability Staining and Debris Exclusion:
- Resuspend the cell pellet in a small volume of Flow Buffer and determine cell count and viability using a hemocytometer or automated counter. Viability should exceed 90% for optimal results [11].
- Add an appropriate viability dye (e.g., 7-AAD) according to manufacturer's instructions and incubate for 5-10 minutes in the dark at 4°C [11].
- Wash cells once with Flow Buffer and resuspend at a final concentration of 0.5-1×10^6 cells/mL in Flow Buffer for acquisition.
Protocol for Resolving and Preventing Instrument Clogs
This troubleshooting protocol provides a systematic approach to address fluidics obstructions, based on established instrument maintenance procedures [59].
Materials Required:
- Deionized Water (DI H₂O)
- Laboratory Detergent: 5-10% Contrad solution or equivalent
- Sodium Hypochlorite: 10% bleach solution
- Wire Stylus (supplied with instrument or available from manufacturer)
- Sample Tubes and appropriate waste container
Clog Resolution Procedure:
- Initial Assessment and Mild Clearing:
- Place a tube with ~1mL of DI H₂O on the sample injection port (SIP).
- Press the Prime button and observe the tube for bubble emission. The presence of a rapid bubble sequence indicates a partial but not complete clog [59].
- Press Run and observe the FSC-A vs. SSC-A plot. If events appear, attempt to run a clean water sample to clear residual debris.
- Moderate Clog Clearing:
- If the system remains clogged, place ~1mL of Contrad solution in a tube and run on "High" for 5 minutes. The detergent helps dissolve organic debris and sticky cells [59].
- Follow with a 1-2 minute DI H₂O run to flush out the detergent and loosened material.
- Attempt to run samples again. If successful, perform a final system flush with DI H₂O.
- Severe Clog Procedure:
- If no bubbles are observed during priming or the system remains completely obstructed, mechanical intervention is required [59].
- Place the instrument in Standby mode and carefully remove the outer SIP tubing by unscrewing the gray knurled nut.
- Using a wire stylus, gently "floss" the inner SIP tubing by inserting the wire and moving it up and down 5-10 cycles to dislodge the obstruction.
- Reassemble the SIP, run Contrad for 5 minutes followed by DI H₂O for 1 minute, then attempt sample acquisition.
- Post-Clog System Sanitization:
- After resolving any clog, especially when working with potentially biohazardous materials, run 10% bleach for 5 minutes to sterilize the fluidics path [60].
- Follow with 5% Contrad soap for 3 minutes and a final DI H₂O rinse for 3 minutes to ensure all cleaning agents are removed from the system.
Data Presentation and Analysis
Quantitative Parameters for Scatter Assessment
The following table summarizes key quantitative metrics for evaluating scatter profile quality, derived from established flow cytometry quality control practices [58] [57].
Table 1: Quantitative Metrics for Scatter Profile Assessment
| Parameter | Optimal Range | Suboptimal Indication | Corrective Action |
|---|---|---|---|
| FSC Peak Coefficient of Variation | <5% | >8% suggests size heterogeneity | Check for clumps; optimize dissociation |
| SSC Peak Width | Tight, defined distribution | Broad distribution indicates granularity variation | Remove dead cells; check fixation method |
| Event Rate | Stable, within 10% of set rate | Fluctuating or declining rate | Check for partial clog; dilute sample |
| % Debris in FSC-low/SSC-low | <10% of total events | >20% of total events | Improve filtration; remove more supernatant |
| Viable Cell Recovery | >85% of initial count | <70% of initial count | Optimize centrifugation; gentle handling |
Research Reagent Solutions for Scatter and Clogging Management
Table 2: Essential Reagents for Managing Scatter and Clogging Issues
| Reagent/Category | Specific Examples | Function and Application |
|---|---|---|
| Enzymatic Detachment Reagents | Trypsin replacement, Accutase | Gentle cell dissociation from substrates to preserve surface markers and scatter properties [56] |
| Viability Dyes | 7-AAD, DAPI, Propidium iodide | Discrimination of dead/dying cells for exclusion during analysis to improve scatter profile clarity [11] |
| Cell Strainers | 40-70μm nylon mesh filters | Removal of cell clumps and large debris before sample acquisition to prevent clogs [58] |
| Blocking Reagents | FcR blocking antibodies, normal serum | Reduction of non-specific antibody binding to improve signal-to-noise in fluorescence detection [30] |
| System Cleaning Solutions | 10% bleach, 5% Contrad detergent | Removal of organic debris and sterilization of fluidics between samples to prevent clogs [59] [60] |
| Fixation/Permeabilization Kits | Commercial kits (e.g., ab185917) | Standardized intracellular antigen access while controlling effects on light scatter properties [11] |
Workflow Visualization
Scatter Optimization and Clog Management Workflow
The following diagram illustrates the integrated procedural pathway for addressing scatter and clogging issues, from sample preparation through data acquisition:
Instrument Clogging Resolution Pathway
This troubleshooting diagram provides a systematic approach to identifying and resolving fluidics obstructions:
Effective management of scatter properties and prevention of instrument clogging are not merely technical exercises but fundamental requirements for generating reliable intracellular stem cell marker data. The protocols and troubleshooting guides presented here provide a systematic framework for addressing these common challenges, emphasizing preventive measures through proper sample handling and regular instrument maintenance. For researchers in drug development and translational science, where quantitative accuracy and reproducibility are paramount, implementing these standardized approaches ensures that critical decisions are based on robust experimental data rather than technical artifacts. As flow cytometry continues to evolve toward higher-parameter panels and more complex cellular analyses, the foundational principles of sample quality and instrument maintenance detailed in this application note will remain essential for scientific rigor and discovery.
Ensuring Reproducibility: Method Validation and Comparative Technology Assessment
Precision, encompassing both intra- and inter-assay reproducibility, is a cornerstone of reliable flow cytometry, especially within the stringent context of intracellular stem cell marker research [61]. For researchers and drug development professionals, demonstrating robust precision is not merely a technical exercise but a fundamental requirement for generating credible, publishable data and for meeting regulatory standards in advanced therapy medicinal product (ATMP) development [62]. Intra-assay precision measures the consistency of results when the same sample is repeatedly analyzed within a single run, reflecting the method's repeatability. In contrast, inter-assay precision evaluates variation across different runs, performed on different days, by different operators, or using different reagent lots, thereby indicating the method's reproducibility [63]. In the analysis of intracellular stem cell markers—where populations can be rare and phenotypic shifts subtle—establishing high precision is critical to distinguish true biological signals from methodological noise, ensuring that conclusions about stem cell purity, differentiation status, and potency are valid [64] [62].
Key Concepts and Calculations
The Coefficient of Variation (%CV) is the standard metric for quantifying precision in flow cytometry. It is a dimensionless number that expresses the standard deviation as a percentage of the mean, allowing for the comparison of variability across different measurements and scales [63]. The calculations for intra- and inter-assay CV are distinct, each providing unique insights into the method's performance.
Intra-Assay CV is calculated from replicate measurements (e.g., duplicates) of each sample within one analytical run. It reflects the immediate repeatability of the assay [63]. For each sample, the %CV is calculated, and the average of these individual %CVs is reported as the intra-assay CV for the experiment.
Inter-Assay CV is calculated from control samples (e.g., high and low controls for a critical marker) run across multiple independent plates or days [63]. First, the mean value for the control on each plate is determined. The overall mean and standard deviation of these plate means are then used to calculate the %CV for each control level. The average of the %CVs from the high and low controls is typically reported as the inter-assay CV.
Table 1: Interpretation of Precision Metrics in Flow Cytometry
| Precision Type | Calculation Basis | Acceptance Criterion (%CV) | Indicates |
|---|---|---|---|
| Intra-Assay | Replicates within a single run [63] | < 10% is desirable [63] | Repeatability (Within-run consistency) |
| Inter-Assay | Control means across multiple runs [63] | < 15% is generally acceptable [63] | Reproducibility (Between-run consistency) |
| Rare Cell Populations | Low-frequency subsets (e.g., <1%) [65] | 30-35% may be acceptable [65] | Assay robustness for rare events |
For rare cell populations, such as specific stem or progenitor cell subsets, higher %CVs are often unavoidable and may be deemed acceptable, with values of 30-35% sometimes permitted due to the inherent statistical challenges in quantifying rare events [65].
Experimental Protocol for Precision Assessment
This protocol outlines a detailed procedure for evaluating the precision of a flow cytometry assay designed to detect an intracellular stem cell transcription factor (e.g., OCT4) in a cultured human pluripotent stem cell line.
Reagent Solutions and Materials
Table 2: Essential Research Reagents and Materials
| Item | Function/Explanation |
|---|---|
| Fc Receptor Blocking Reagent [9] [11] | Prevents non-specific antibody binding via Fc receptors, reducing background noise. |
| Fixation/Permeabilization Kit [11] | Preserves cell structure (fixation) and renders membranes permeable to antibodies (permeabilization) for intracellular staining. |
| Fluorochrome-Conjugated Antibodies | Target-specific antibodies for surface (e.g., TRA-1-60) and intracellular (e.g., OCT4) markers. Titration is critical [66]. |
| Isotype Control Antibodies [9] [66] | Matched to the primary antibodies, they distinguish non-specific binding from specific signal. |
| Viability Dye (e.g., 7-AAD) [62] [11] | Identifies and allows for the exclusion of dead cells, which are prone to non-specific antibody uptake. |
| Flow Cytometry Staining Buffer [9] | A buffer (e.g., PBS with BSA) for washing and resuspending cells, preserving viability and reducing non-specific staining. |
| Counting Beads [62] | Synthetic beads of known concentration used as a surrogate material to qualify cell enumeration methods. |
Staining and Data Acquisition Workflow
The following diagram illustrates the core experimental workflow for preparing and analyzing cells for intracellular marker detection.
Step-by-Step Procedure
- Sample Preparation: Harvest human pluripotent stem cells using a gentle cell dissociation reagent to preserve surface antigens. Avoid trypsin if possible, as it can cleave surface markers; if required, allow a recovery period of 6-10 hours post-trypsinization [9]. Wash cells three times in cold PBS supplemented with 0.5% BSA by centrifugation at 350–500 × g for 5 minutes [9]. Determine total cell count and ensure viability exceeds 90% [11].
- Viability Staining: Resuspend the cell pellet in cold buffer and stain with a viability dye (e.g., 7-AAD) according to the manufacturer's protocol. Incubate in the dark at 4°C. Wash cells twice to remove unbound dye [11].
- Fc Receptor Blocking: Resuspend the cell pellet in an Fc receptor blocking buffer (e.g., 2-10% goat serum or human IgG) and incubate for 30-60 minutes in the dark at 4°C [9] [11]. Do not wash after this step [9].
- Surface Marker Staining: Directly add a titrated, fluorochrome-conjugated antibody against a surface stem cell marker (e.g., anti-TRA-1-60) to the cells. Vortex gently and incubate for 30 minutes in the dark on ice [9] [66]. Wash the cells twice with 2 mL of cold staining buffer to remove unbound antibody.
- Fixation and Permeabilization: Fix cells by resuspending the pellet in 1-4% paraformaldehyde (PFA) and incubating for 15-20 minutes on ice [11]. Wash twice with suspension buffer. Permeabilize cells by resuspending in a mild detergent solution (e.g., 0.2% Saponin in PBS) and incubating for 10-15 minutes at room temperature [11].
- Intracellular Staining: Add a titrated, fluorochrome-conjugated antibody against the intracellular target (e.g., anti-OCT4) to the cell pellet in permeabilization buffer. Incubate for 30 minutes in the dark at room temperature. Wash the cells twice with permeabilization buffer, followed by a final wash with standard staining buffer.
- Data Acquisition: Resuspend the final cell pellet in 200–400 µL of staining buffer [9]. Filter the cell suspension through a FACS tube mesh before acquiring data on the flow cytometer [66]. Acquire a minimum of 10^5 total events per sample to ensure robust statistics, particularly for rare subsets [65]. For the precision study, prepare and analyze the same sample in triplicate within the same run (for intra-assay CV) and again across three separate runs on different days (for inter-assay CV) [65].
Validation Parameters and Acceptance Criteria
A comprehensive precision assessment is part of a broader assay validation or qualification framework, which is essential for regulatory compliance in drug development [62] [61]. The following diagram and table outline the key parameters of this framework.
Table 3: Key Parameters for Flow Cytometry Assay Validation
| Parameter | Definition | Evaluation in Precision Context |
|---|---|---|
| Precision [61] | The closeness of agreement between a series of measurements. | Quantified via Intra-assay CV (repeatability) and Inter-assay CV (reproducibility) as described [63]. |
| Accuracy [61] | The closeness of the measured value to the true value. | Assessed by comparing flow cytometry results with a known standard or reference method. |
| Specificity [61] | The ability to measure specifically the target analyte. | Demonstrated through proper gating on viable, single cells and use of isotype/full stain controls [11] [66]. |
| Robustness [61] | The capacity to remain unaffected by small, deliberate variations in method parameters. | Tested by evaluating precision and accuracy when altering incubation times (±5 min), temperature, or antibody volumes. |
| Linearity & Range [61] | The interval over which the analytical performance is maintained. | Determined by staining a dilution series of cells and ensuring the %CV remains acceptable across the expected sample concentration range. |
For a potency assay on a Natural Killer (NK) cell product, a Phase I/IIa study established acceptance criteria for its flow cytometry-based method prior to validation, ensuring it was "fit-for-purpose" [62]. This approach is directly applicable to stem cell therapy development.
Accurately determining the sensitivity of a flow cytometry assay is a critical step in protocol validation, defining the smallest quantity of a target that can be reliably detected and measured. For researchers investigating intracellular stem cell markers, establishing a rigorously defined Lower Limit of Detection (LLoD) and Lower Limit of Quantification (LLoQ) is paramount. These parameters ensure that observed variations in pluripotency markers such as NANOG or other undifferentiated stem cell markers genuinely reflect biological changes rather than assay noise, enabling confident detection of rare cell populations and subtle phenotypic shifts essential in drug development [67] [13].
This application note details the experimental protocols and data analysis frameworks for establishing these crucial sensitivity parameters within the context of intracellular stem cell marker research. The principles outlined are also broadly applicable to other high-sensitivity flow cytometry applications, including measurable residual disease (MRD) detection in oncology and soluble biomarker analysis [68] [69] [70].
Theoretical Foundations of Assay Sensitivity
The sensitivity of a flow cytometry assay is formally characterized by two key parameters: the Lower Limit of Detection (LLoD) and the Lower Limit of Quantification (LLoQ).
- Lower Limit of Detection (LLoD): The LLoD represents the lowest concentration of an analyte that can be reliably distinguished from a blank sample. It is a binary measure of presence or absence. Statistically, it is often derived as the concentration corresponding to a signal intensity that is three standard deviations above the mean of a zero-concentration (blank) sample.
- Lower Limit of Quantification (LLoQ): The LLoQ is the lowest analyte concentration that can be quantitatively measured with acceptable precision and accuracy, typically defined by a coefficient of variation (CV) of ≤20-25% [67]. The LLoQ is inherently linked to the number of cellular events acquired during analysis. Acquiring more events increases the statistical power to identify and precisely measure rare cell populations [67].
For instance, in MRD detection, achieving an LLoQ of 0.01% (10⁻⁴) requires the acquisition of at least 500,000 leukocyte events to confidently identify a cluster of 50 abnormal cells. Acquiring 5 million events can theoretically lower the LLoQ to 0.001% (10⁻⁵), dramatically enhancing the assay's ability to detect minimal disease [67] [69]. This principle is directly transferable to stem cell research, where identifying rare sub-populations based on intracellular marker expression is often the goal.
The following diagram illustrates the core relationship between event acquisition and sensitivity thresholds, which is fundamental to experimental design in high-sensitivity flow cytometry.
Experimental Protocols for Sensitivity Determination
Protocol 1: Determining LLoD for an Intracellular Stem Cell Marker
This protocol outlines the procedure for establishing the Lower Limit of Detection for an intracellular target, such as the pluripotency marker NANOG, in human induced pluripotent stem cells (iPSCs) [13].
Materials:
- Human iPSC culture
- Validated antibody against target intracellular antigen (e.g., anti-NANOG)
- Isotype control antibody
- Flow cytometry staining buffer (PBS with 0.5-1% BSA)
- Fixation solution (e.g., 1-4% Paraformaldehyde (PFA))
- Permeabilization solution (e.g., 0.1-0.5% Triton X-100, Saponin, or 90% Methanol)
- Fc receptor blocking reagent
- Round-bottom FACS tubes
- Centrifuge and vortex mixer
- Flow cytometer (e.g., BD FACSLyric)
Method:
- Cell Preparation and Fixation: Harvest and wash a homogeneous culture of iPSCs. Create a single-cell suspension and determine viable cell count and viability, which should ideally be 90-95% [11]. Split the cells into multiple aliquots.
- Sample Serial Dilution: Serially dilute the iPSCs with a known high expression of the target marker into a background of unstained, fixed, and permeabilized iPSCs that are negative for the marker. This creates a dilution series spanning expected low abundance levels (e.g., 1%, 0.1%, 0.01%, 0.001%).
- Intracellular Staining: Following fixation and permeabilization optimized for your target antigen, stain all sample aliquots with the target-specific antibody and appropriate isotype controls [71] [13]. A suggested fixation and permeabilization method is below.
- Fixation: Resuspend cell pellet in 100 µL of ice-cold 4% PFA. Incubate for 20 minutes at room temperature. Wash with 2 mL of staining buffer [71].
- Permeabilization: Resuspend fixed cell pellet in 100 µL of permeabilization solution (e.g., 0.1% Triton X-100 in PBS). Incubate for 10-15 minutes at room temperature. Wash with 2 mL of staining buffer [71].
- Antibody Incubation: Resuspend cells in staining buffer containing the pre-titrated antibody. Incubate for 30 minutes in the dark. Wash twice with staining buffer before acquisition [9].
- Data Acquisition: Acquire a sufficiently high number of total events for each dilution sample, targeting a minimum of 1-2 million events per tube to ensure robust statistical analysis of very low-frequency populations [67] [69].
- Data Analysis: For each dilution, identify the positive population and record the event count. The LLoD is statistically derived from the dilution where the positive population can be distinguished from the negative control with 95% confidence. A common approach is to use the formula: LLoD = Mean(Blank) + 3 * SD(Blank), where the "Blank" is the negative control sample.
Protocol 2: Determining LLoQ for an Intracellular Stem Cell Marker
This protocol builds upon the LLoD determination to establish the lowest concentration that can be measured with quantitative accuracy.
Materials: (As per Protocol 1)
Method:
- Preparation of Precision Samples: Prepare a minimum of five replicates of samples at each concentration level around the estimated LLoD (e.g., 0.01%, 0.02%, 0.05%) from the serial dilution series.
- Sample Staining and Acquisition: Stain and acquire all replicate samples in an identical manner, following the intracellular staining protocol detailed in Protocol 1. Ensure consistent instrument settings and acquisition volumes across all runs.
- Inter-Assay Precision (Optional): To assess inter-assay precision, repeat the entire experiment (staining and acquisition) over five separate days [68].
- Data Analysis: Calculate the mean measured concentration and the Coefficient of Variation (CV = [Standard Deviation / Mean] * 100%) for the replicates at each concentration level. The LLoQ is defined as the lowest concentration level at which the CV is ≤20% (or another pre-defined acceptability threshold, e.g., 25%) [67].
Data Analysis and Interpretation
The data collected from the above protocols must be systematically analyzed to formally establish the LLoD and LLoQ. The following table summarizes quantitative sensitivity data from various flow cytometry applications, illustrating achievable benchmarks.
Table 1: Exemplary Sensitivity Parameters from Flow Cytometry Assays
| Application / Target | Reported LLoD | Reported LLoQ / Sensitivity | Key Experimental Parameters |
|---|---|---|---|
| Soluble Biomarkers (sCD25, sTREM-1) [68] | 9.77 pg/mL (sCD25) 12.21 pg/mL (sTREM-1) | Not specified | Bead-based immunoassay; >10,000 events recorded. |
| AML MRD Detection [70] | Not specified | 0.01% | 10-color panel; 1-1.2 million events/tube; 3-tube panel. |
| High-Sensitivity MRD [67] [69] | 50 cells (Theoretical LLoD) | 0.001% - 0.01% | >500,000 to 5 million acquired events. |
After calculating the CV% for each concentration level from Protocol 2, the results should be plotted to visually determine the LLoQ.
Table 2: Example Data Analysis for LLoQ Determination of a Hypothetical Stem Cell Marker
| Theoretical Concentration (%) | Mean Measured Concentration (%) | Standard Deviation | CV (%) | Meets CV ≤20% Criterion? |
|---|---|---|---|---|
| 0.001 | 0.0012 | 0.0005 | 41.7 | No |
| 0.005 | 0.0053 | 0.0015 | 28.3 | No |
| 0.010 | 0.0098 | 0.0020 | 20.4 | Yes (LLoQ) |
| 0.050 | 0.048 | 0.006 | 12.5 | Yes |
The workflow for analyzing the acquired data to establish both limits is summarized below, connecting the key steps of gating, statistical analysis, and final parameter determination.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful implementation of high-sensitivity intracellular flow cytometry relies on a carefully selected set of reagents and tools.
Table 3: Essential Reagents and Materials for Intracellular Assay Development
| Reagent / Material | Function / Purpose | Examples & Considerations |
|---|---|---|
| Fixation Solution | Preserves cellular structure and cross-links intracellular proteins, locking them in place. | 1-4% Paraformaldehyde (PFA); crosslinking fixatives are preferable for post-translational modifications [71] [14]. |
| Permeabilization Reagent | Disrupts the cell membrane to allow antibodies access to intracellular targets. | Detergents like Triton X-100 (strong, for nuclear targets), Saponin (mild, reversible), or Methanol (standalone fix & perm) [11] [71]. |
| Validated Primary Antibodies | Specifically bind to the intracellular antigen of interest. | Antibodies validated for flow cytometry and compatible with the chosen fix/perm method. Titration is essential [13]. |
| Fc Receptor Blocking Reagent | Prevents non-specific antibody binding via Fc receptors, reducing background noise. | Human IgG, mouse anti-CD16/CD32, or serum from an unrelated species [11] [9]. |
| Flow Cytometry Staining Buffer | Provides an isotonic medium for washing and antibody dilution. | PBS supplemented with 0.5-1% BSA or FCS; may contain sodium azide [9]. |
| High-Sensitivity Flow Cytometer | Instrument for detecting and quantifying fluorescence at the single-cell level. | Systems like BD FACSLyric [68] [70] with stable lasers and sensitive detectors are critical for low-abundance targets. |
Establishing a rigorously defined LLoD and LLoQ is a non-negotiable component of assay validation for intracellular stem cell marker research. The protocols detailed herein provide a framework for determining these parameters, emphasizing the importance of robust statistical analysis, adequate event acquisition, and controlled experimental conditions. By formally characterizing assay sensitivity, researchers and drug development professionals can generate reliable, reproducible, and clinically or scientifically meaningful data, ultimately advancing the field of stem cell biology and therapy.
Correlating Flow Cytometry Data with Other Pluripotency Assays
Within stem cell research and drug development, the accurate assessment of pluripotency—the capacity of a cell to differentiate into all somatic cell lineages—is paramount. Flow cytometry has become an indispensable tool for this purpose, allowing for the quantitative analysis of intracellular pluripotency markers at the single-cell level. However, the full power of flow cytometric data is only realized when it is correlated with other, functionally distinct pluripotency assays. This integrated approach provides a more comprehensive and reliable validation of a cell's pluripotent state, moving beyond mere marker expression to confirm functional potential. This application note details protocols for intracellular staining of stem cell markers and provides a framework for correlating this data with key alternative assays, thereby strengthening conclusions in research and development workflows.
Experimental Design and Quantitative Data Correlation
Successful correlation begins with a robust experimental plan that incorporates multiple assay types. Key assays to run in parallel with flow cytometry are summarized in the table below.
Table 1: Key Pluripotency Assays for Correlation with Flow Cytometry Data
| Assay Type | Measured Parameter | Key Outputs | Correlation with Flow Cytometry |
|---|---|---|---|
| Immunocytochemistry (ICC) | Protein expression and sub-cellular localization | Qualitative/ Semi-quantitative imaging of markers (e.g., OCT4, NANOG) | Confirms staining specificity and reveals heterogeneity in the expression levels and nuclear localization of key markers within the population [56]. |
| Quantitative PCR (qPCR) | Gene expression levels | mRNA expression fold-change of pluripotency genes (e.g., POUSF1, SOX2, NANOG) | Provides a bulk population measurement that should align with the protein-level data; high flow positivity should correlate with high mRNA expression [56]. |
| Embryoid Body (EB) Formation | Spontaneous differentiation capacity | Formation of 3D aggregates; differentiation into cell types of the three germ layers | A functionally positive population in flow cytometry (high for pluripotency markers) should demonstrate high efficiency in EB formation and subsequent multi-lineage differentiation. |
| Teratoma Formation | In vivo differentiation potential | Formation of a complex tissue containing derivatives of ectoderm, mesoderm, and endoderm | The gold-standard assay; cells identified as pluripotent by flow cytometry should be capable of forming teratomas, validating the in vivo functional relevance of the marker profile. |
The following workflow diagram illustrates the strategic integration of these assays in a typical experimental plan.
Detailed Protocol: Intracellular Staining for Pluripotency Markers by Flow Cytometry
This protocol is optimized for the detection of nuclear transcription factors critical for pluripotency, such as OCT4, SOX2, and NANOG, while allowing for simultaneous analysis of surface markers [26].
Materials and Reagents
Table 2: Essential Research Reagent Solutions for Intracellular Staining
| Item | Function | Specific Examples & Notes |
|---|---|---|
| Foxp3/Transcription Factor Staining Buffer Set | Combined fixation/permeabilization solution optimized for nuclear antigens [26]. | Thermo Fisher Scientific (cat. no. 00-5523) |
| Flow Cytometry Staining Buffer | Buffer for washing and resuspending cells; contains protein to reduce background [26]. | PBS with 0.5-1% BSA or 2-5% FBS [26] [11]. |
| Fixable Viability Dye (FVD) | Distinguishes live from dead cells to exclude the latter from analysis, crucial for accuracy [26] [11]. | eFluor 450, 506, 780; choose a dye not overlapping with antibody fluorochromes [26]. |
| Directly Conjugated Antibodies | Detection of specific surface and intracellular targets. | Validated antibodies against OCT4-Alexa Fluor 488, SOX2-PE, SSEA-1-APC, etc. |
| Fc Receptor Blocking Reagent | Reduces non-specific antibody binding [11]. | Normal serum (e.g., mouse, rat) or purified CD16/CD32 antibody [26] [11]. |
Step-by-Step Methodology
1. Sample Preparation and Viability Staining
- Harvest cells to create a single-cell suspension using a gentle method like Accutase [56].
- Wash cells with cold staining buffer by centrifuging at 200-400 x g for 5 minutes at 4°C [11].
- Resuspend cell pellet in PBS or buffer at a concentration of 0.5–1 x 10⁶ cells/mL [11].
- Optional but recommended: Stain cells with a fixable viability dye according to the manufacturer's instructions. Wash cells twice post-staining to remove unbound dye [26] [11].
2. Cell Surface Staining (for co-analysis with surface markers)
- Resuspend the cell pellet in 50-100 µL of staining buffer.
- Add directly conjugated antibodies against surface antigens (e.g., CD324, SSEA-1, TRA-1-60) at the predetermined optimal concentration.
- Incubate for 20-30 minutes on ice or at 4°C, protected from light.
- Wash cells twice with 2 mL of staining buffer to remove unbound antibody [26] [56].
3. Fixation and Permeabilization for Intracellular Antigens
- After the final surface stain wash, thoroughly resuspend the cell pellet in the residual buffer.
- Fix and permeabilize cells by adding 1 mL of freshly prepared Foxp3 Fixation/Permeabilization working solution per sample. Mix immediately by vortexing.
- Incubate for 30-60 minutes at room temperature, protected from light [26].
- Add 2 mL of 1X Permeabilization Buffer and centrifuge at 400-600 x g for 5 minutes. Discard the supernatant. Repeat this wash step once [26].
4. Intracellular Staining
- Resuspend the fixed and permeabilized cell pellet in 100 µL of 1X Permeabilization Buffer.
- Optional Fc Block: Add 2 µL of normal serum to the cell suspension and incubate for 15 minutes at room temperature [26].
- Without washing, add the recommended amount of directly conjugated antibodies against intracellular targets (e.g., OCT4, SOX2, NANOG).
- Incubate for 30-60 minutes at room temperature, protected from light [26].
- Wash cells twice with 2 mL of 1X Permeabilization Buffer.
- Resuspend the final cell pellet in an appropriate volume of Flow Cytometry Staining Buffer for analysis on the flow cytometer [26].
Critical Controls and Data Acquisition
- Controls are essential: Include unstained cells, fluorescence-minus-one (FMO) controls for each fluorochrome, and isotype controls where appropriate to set gates and determine positive populations [72].
- Instrument Setup: Perform daily quality control on the flow cytometer. Use compensation controls (e.g., antibody capture beads or stained cells) to correct for spectral overlap between fluorochromes [72].
- Acquisition: Collect a statistically significant number of events (e.g., >10,000 events for the population of interest). Record all cells initially and use a sequential gating strategy during analysis to exclude debris, doublets, and dead cells [20] [72].
The following diagram outlines the key steps and decision points in the protocol.
Data Interpretation and Correlation Strategy
The correlation of data across different assays strengthens the final conclusion about the pluripotent state of the cells.
- Flow Cytometry and ICC/qPCR: Flow cytometry provides a quantitative percentage of positive cells and their fluorescence intensity distribution. This data should be directly comparable to the qualitative (but spatially resolved) protein expression in ICC and the quantitative mRNA levels from qPCR. A strong, positive population in flow cytometry should be reflected by intense nuclear staining in ICC and high Ct values in qPCR for the corresponding genes [56].
- Flow Cytometry and Functional Assays (EB/Teratoma): The most critical correlation is between marker expression and function. The percentage of cells highly positive for core pluripotency markers by flow cytometry should predict the efficiency of the culture to form EBs. For teratoma assays, a high degree of purity of the pluripotent population, as determined by flow cytometry, is typically required for consistent and rapid teratoma formation in vivo.
In conclusion, a multi-faceted approach that correlates quantitative flow cytometry data with other pluripotency assays provides the most robust framework for validating stem cell status. The protocols and correlation strategies detailed herein offer researchers a concrete pathway to generate reliable, reproducible, and publication-quality data for both basic research and drug development applications.
Flow cytometry stands as a critical technology in modern biological research and drug development, enabling multiparameter analysis at the single-cell level. For researchers investigating intracellular stem cell markers, choosing between conventional and spectral flow cytometry represents a significant strategic decision that profoundly impacts experimental design, panel complexity, and data quality. This comparative analysis examines both technologies within the context of high-parameter panel development for stem cell research, providing detailed technical specifications, experimental protocols, and practical implementation guidelines to inform instrument selection and methodological approach.
Technical Fundamentals Comparison
The fundamental distinction between conventional and spectral flow cytometry lies in their approach to detecting and resolving fluorescent signals. Conventional flow cytometry operates on a principle of optical filtering and compensation, where emitted light from fluorophores is directed through a series of mirrors and bandpass filters to discrete detectors, typically with one primary detector assigned to each fluorophore [73]. This system requires mathematical "compensation" to subtract spectral spillover between channels, wherein a portion of a fluorophore's emission spectrum is detected in a channel primarily assigned to another fluorophore [74]. This approach inherently limits the number of parameters that can be simultaneously resolved to the number of available detectors, with current practical limits typically reaching 15-20 colors [73].
In contrast, spectral flow cytometry captures the complete emission spectrum of every fluorophore across a broad wavelength range (approximately 350-900 nm) using an array of detectors [75]. Rather than assigning fluorophores to primary channels, spectral instruments employ "unmixing" algorithms that utilize the full spectral signature of each fluorophore to distinguish multiple markers within a sample [73] [75]. This holistic approach enables several key advantages: it allows resolution of fluorophores with nearly identical peak emissions but distinct off-peak patterns, facilitates autofluorescence extraction to improve signal resolution, and dramatically expands parameter capacity, with panels now exceeding 40 colors demonstrated in research applications [76] [75].
Table 1: Technical Comparison of Conventional and Spectral Flow Cytometry
| Feature | Conventional Flow Cytometry | Spectral Flow Cytometry |
|---|---|---|
| Detection Method | Bandpass filters; single detector per fluorophore | Full spectrum collection; multiple detectors per fluorophore |
| Spillover Correction | Compensation (mathematical subtraction) | Unmixing (spectral signature recognition) |
| Typical Parameter Limit | 15-20 colors [73] | 40+ colors [76] [75] |
| Wavelength Range | Narrow bands near emission maxima | Broad spectrum (350-900 nm) [75] |
| Autofluorescence Handling | Limited correction capability | Can be extracted as separate parameter [75] [74] |
| Fluorophore Selection Constraints | Limited by filter configuration and spillover | Limited by spectral signature uniqueness [75] |
Diagram 1: Fundamental workflow comparison between conventional and spectral flow cytometry technologies.
Experimental Protocols for Intracellular Stem Cell Marker Analysis
Sample Preparation and Staining Protocol
The following protocol has been optimized for intracellular stem cell marker analysis, incorporating critical steps to preserve epitope integrity and ensure specific staining:
Cell Harvesting: For adherent stem cell cultures, gently wash with Mg²⁺/Ca²⁺-free PBS at room temperature. Use pre-warmed trypsin replacement or Accutase to detach cells, incubating at 37°C for 2-5 minutes. Avoid over-digestion, which can compromise surface epitopes and cell viability [56]. Quench enzymatic activity with twice the volume of flow buffer (2% FBS in PBS) and collect cells in a 15 mL conical tube.
Fixation and Permeabilization: Centrifuge cell suspension at 300 × g for 5 minutes and resuspend in 4% paraformaldehyde for 15 minutes at room temperature to fix cells. Wash twice with flow buffer, then resuspend in ice-cold 90% methanol for 30 minutes on ice for permeabilization. Alternative permeabilization buffers (e.g., saponin-based) may be optimized for specific intracellular epitopes [56].
Intracellular Staining: Wash methanol-permeabilized cells twice with flow buffer. Incubate with fluorophore-conjugated primary antibodies targeting intracellular stem cell markers (e.g., transcription factors) for 30 minutes at room temperature protected from light. Optimal antibody concentrations should be determined through titration for each new antibody lot [56].
Critical Controls: Include unstained cells for autofluorescence assessment, fluorescence-minus-one (FMO) controls for gate setting, and single-stained compensation controls for each fluorophore. For spectral cytometry, single-stained controls are essential for establishing reference spectral signatures [74]. Compensation beads can be used for abundant markers, but cells are preferred for rare markers to ensure accurate spectral signature capture [74].
Reference Control Preparation for Spectral Unmixing
Spectral flow cytometry requires meticulous preparation of reference controls to establish the unique spectral signature for each fluorophore used in the panel:
Single-Stain Controls: Prepare individual samples stained with each fluorophore-conjugated antibody used in the full panel. Use the same cell type as experimental samples when possible, as autofluorescence properties are cell-type specific [74].
Batch Consistency: Ensure single-stain controls and full-panel samples are stained with antibodies from the same manufacturing lot, particularly for tandem dyes (e.g., PE-Cy7), which exhibit batch-to-batch variation in spectral properties [74].
Unstained Control: Include an unstained sample of the same cell type to establish baseline autofluorescence, which can be computationally extracted during unmixing to improve signal resolution [75] [74].
Panel Design Strategies for High-Parameter Applications
Fluorophore Selection and Assignment
Effective high-parameter panel design requires strategic fluorophore selection and assignment based on marker expression patterns and fluorophore properties:
Brightness-Antigen Matching: Assign brighter fluorophores (e.g., PE, APC) to lowly expressed antigens, while dimmer fluorophores should be paired with highly expressed antigens to optimize resolution and minimize spillover spreading error [76] [74]. For stem cell applications, bright fluorophores are particularly important for detecting transcription factors with relatively low expression levels.
Spectral Spacing: In spectral cytometry, prioritize fluorophores with distinct spectral signatures, especially for co-expressed markers. Utilize spectral viewing tools (e.g., Cytek's Similarity Index) to assess overlap between fluorophores, aiming for combinations with low similarity scores (<0.3) for markers expressed on the same cell population [74].
Laser Utilization: Distribute fluorophores across available laser lines (UV, violet, blue, yellow-green, red) to minimize excitation overlap. Modern spectral cytometers typically feature 5-6 lasers, enabling comprehensive coverage of the fluorescence spectrum [76] [75].
Table 2: Essential Research Reagent Solutions for Intracellular Stem Cell Marker Analysis
| Reagent Category | Specific Examples | Function in Experimental Protocol |
|---|---|---|
| Dissociation Reagents | Trypsin replacement, Accutase | Generates single-cell suspension from adherent cultures [56] |
| Fixation/Permeabilization | Paraformaldehyde, Methanol, Saponin-based buffers | Preserves cellular structure and enables antibody access to intracellular targets [56] |
| Flow Buffer | PBS with 2% FBS | Provides isotonic suspension medium while reducing non-specific antibody binding |
| Viability Markers | 7-AAD, Propidium iodide | Distinguishes live from dead cells to improve analysis accuracy [77] |
| Antibody Panels | CD markers, transcription factor antibodies | Enables specific detection of stem cell populations and differentiation states [10] |
| Reference Controls | Compensation beads, single-stain antibodies | Establishes spectral references for unmixing algorithms [74] |
| Intracellular Staining Antibodies | Anti-FoxP3, Anti-Nanog, Anti-Oct4 | Targets key stem cell transcription factors for pluripotency assessment [77] [10] |
Implementation of a 33-Color Immunophenotyping Panel
A recently published 33-color spectral flow cytometry panel for comprehensive immune cell characterization demonstrates effective implementation of these principles [76]. This panel employs careful laser and fluorophore distribution across five laser lines (UV, violet, blue, yellow-green, red), with bright fluorophores assigned to low-abundance markers and dimmer fluorophores to highly expressed antigens. The panel successfully characterizes T-cell, B-cell, NK-cell, and dendritic cell subpopulations in peripheral blood, demonstrating the practical feasibility of high-parameter analysis. For stem cell researchers, this approach can be adapted to simultaneously examine pluripotency markers, differentiation antigens, and functional state indicators in complex cultures.
Diagram 2: Systematic workflow for designing high-parameter flow cytometry panels, emphasizing iterative optimization.
Analytical Approaches and Data Interpretation
Unmixing Algorithms and Computational Analysis
Spectral flow cytometry data analysis relies on sophisticated unmixing algorithms that mathematically separate the contributions of multiple fluorophores from the composite spectrum detected for each cell. These algorithms, including principal component analysis and least squares unmixing, compare the detected signal against reference spectra from single-stain controls to calculate the proportionate contribution of each fluorophore [73]. This approach enables several analytical advantages beyond conventional compensation:
Autofluorescence Extraction: Cellular autofluorescence can be treated as an additional spectral signature and computationally separated from specific antibody-derived signals, significantly improving resolution for dimly expressed markers [75] [74]. This is particularly valuable in stem cell research where autofluorescence can vary between differentiation states.
Similarity Index Assessment: Fluorophore pairs can be evaluated using similarity indices (ranging from 0-1), with lower values indicating more distinct spectral signatures. For optimal resolution, markers with high cellular co-expression should be paired with fluorophores possessing low similarity indices (<0.3) [74].
High-Dimensional Analysis: The increased parameter capacity enables comprehensive immunophenotyping and detection of rare cell populations. For stem cell applications, this allows simultaneous assessment of pluripotency markers, lineage commitment indicators, and functional state proteins within heterogeneous cultures [10].
Application to Stem Cell Research
Flow cytometry has become indispensable for stem cell characterization, enabling identification of rare populations through specific marker combinations. Key applications include:
Pluripotency Assessment: Simultaneous detection of intracellular transcription factors (e.g., Nanog, Oct4, Sox2) and surface markers (e.g., CD133, CD34) provides comprehensive pluripotency evaluation [10]. Spectral cytometry enables expanded panels that incorporate additional functional markers without sacrificing resolution.
Lineage Commitment Analysis: Tracking downregulation of pluripotency markers alongside emergence of lineage-specific markers during differentiation enables detailed mapping of developmental trajectories [10] [56]. High-parameter panels allow capture of multiple lineage branches within single experiments.
Cell Cycle and Functional Status: Incorporation of DNA dyes (e.g., 7-AAD) and metabolic indicators provides additional layers of functional information alongside phenotypic characterization [10].
Technology Selection Guidelines
The choice between conventional and spectral flow cytometry depends on multiple experimental factors:
Panel Complexity: Conventional systems suffice for panels up to 15-20 parameters, while spectral systems excel with 20+ parameters, particularly when incorporating fluorophores with overlapping emission spectra [73] [75].
Cell Population Rarity: Spectral cytometry's autofluorescence extraction provides superior resolution for rare cell detection, a critical consideration for stem cell researchers investigating minor subpopulations [75] [74].
Experimental Throughput: Both technologies support high-throughput applications with plate loader capabilities, though conventional systems may offer faster analysis times for simpler panels [73].
Resource Considerations: Conventional cytometers represent a lower initial investment and may be preferable for laboratories with established panels that don't require expansion. Spectral systems offer greater future-proofing for laboratories anticipating increasing panel complexity [78].
For stem cell researchers specifically focused on intracellular marker analysis, spectral cytometry provides distinct advantages for resolving complex combinations of transcription factors and signaling molecules while extracting interfering autofluorescence. The technology's expanding parameter capacity enables more comprehensive stem cell characterization within increasingly complex differentiation systems, including organoid models [10].
Benchmarking and Standardization for Multi-Laboratory Reproducibility
Reproducibility in flow cytometry data, particularly in the analysis of intracellular stem cell markers, is a significant challenge in multi-laboratory settings. Variations in sample preparation, instrument configuration, and data analysis can lead to inconsistent results, undermining the validity and comparability of research findings. This application note provides a standardized framework and detailed protocols to enhance reproducibility, focusing on the characterization of induced pluripotent stem cells (iPSCs). The procedures outlined are designed to be robust and transferable across different laboratory environments, which is critical for collaborative research and drug development.
Experimental Design and Marker Selection
A foundational step for ensuring reproducibility is the careful selection and pairing of pluripotency markers. Key markers for iPSC characterization are located in specific cellular compartments, which directly influences staining protocols and the choice of antibodies.
Table 1: Common Pluripotency Markers for iPSC Characterization [79]
| Marker | Cellular Location | Primary Application |
|---|---|---|
| OCT4 | Intracellular | Pluripotency verification |
| NANOG | Intracellular | Pluripotency verification |
| SOX2 | Intracellular | Pluripotency verification |
| TRA-1-60 | Surface | Pluripotency verification |
| SSEA-4 | Surface | Pluripotency verification |
For simultaneous staining of two markers (double staining), it is recommended to pair one intracellular marker with one surface marker. This strategy, combined with primary antibodies from different species (e.g., rabbit and mouse) and secondary antibodies conjugated to distinctly colored fluorophores, allows for clearer differentiation under the microscope [79].
Standardized Staining Protocol for Intracellular and Surface Markers
The following is a consolidated and optimized protocol for the staining of intracellular and surface markers in iPSCs, suitable for flow cytometry analysis. This protocol is designed to minimize cell loss and preserve epitope integrity.
Cell Preparation and Fixation
- Cell Culture and Harvesting: Culture iPSCs to 30-40% confluency. Use tissue-culture-treated plates, such as Falcon plates from Corning (Cat #353047), potentially with a pre-coating of iMatrix 511 [79]. For flow cytometry, harvest cells into a single-cell suspension using standard methods [13].
- Fixation: Fix cells by adding 4% paraformaldehyde (PFA) in PBS and incubating for 20 minutes at room temperature [79].
- Permeabilization: For intracellular markers, aspirate the PFA, wash with PBS, and add 0.2% Triton-X-100 in PBS. Incubate for 10 minutes at room temperature. This step is essential for enabling antibodies to access intracellular targets [79].
- Blocking: Add an appropriate blocking buffer (e.g., 10% serum) and incubate for 1 hour at room temperature to reduce non-specific antibody binding [79].
Antibody Staining
A simultaneous staining method after fixation and permeabilization is recommended to reduce cell loss from repeated washing steps. This approach has been validated to yield comparable results to traditional serial staining while improving efficiency [14].
- Antibody Incubation: Dilute primary antibodies against the target surface and intracellular markers in blocking buffer. Add the antibody mixture to the cells and incubate at 4°C for at least one hour (overnight incubation is often recommended) [79].
- Secondary Antibody Staining: If using a purified primary antibody, dilute fluorophore-conjugated secondary antibodies in blocking buffer. Add to the cells and incubate at room temperature for one hour, protecting the plate from light [79].
- Nuclear Staining (Optional): For immunocytochemistry, a DAPI stain (2 µg/ml) can be applied for 10 minutes to visualize cell nuclei [79].
- Acquisition: Resuspend cells in an appropriate buffer for flow cytometry acquisition. Follow instrument-specific protocols for setup and quality control [13].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Intracellular Flow Cytometry [79] [14] [13]
| Reagent | Function | Example |
|---|---|---|
| Fixative | Preserves cell morphology and protein structure | 4% Paraformaldehyde (PFA) |
| Permeabilization Agent | Creates pores in the cell membrane for antibody access | 0.2% Triton X-100 |
| Blocking Buffer | Reduces non-specific antibody binding | Serum (e.g., 10% goat serum) |
| Primary Antibodies | Bind specifically to target antigens (e.g., OCT4, NANOG) | Mouse anti-OCT4, Rabbit anti-NANOG |
| Secondary Antibodies | Fluorophore-conjugated antibodies for detection | Anti-mouse IgG-Alexa Fluor 488 |
| Viability Dye | Distinguishes live from dead cells | Not specified in results, but standard practice [80] |
Optimization and Troubleshooting
Even with standardized protocols, optimization is often required to address common issues such as high background or weak signals.
Table 3: Troubleshooting Common Staining Issues [79]
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| High Background Signal | Non-specific antibody binding | Increase number or duration of washes; Increase blocking buffer concentration (e.g., 10% to 15%); Reduce antibody concentration; Switch to monoclonal antibodies to reduce cross-reactivity [79]. |
| Weak Immunofluorescence Signal | Low antibody binding or detection | Reduce washes; Reduce blocking buffer concentration or switch agent; Increase antibody concentration; Verify permeabilization step for intracellular markers; Confirm microscope filter compatibility [79]. |
Data Standardization and Quality Control
Robust quality control measures are non-negotiable for multi-laboratory reproducibility.
- Instrument Calibration: Regularly calibrate flow cytometers using standard beads. Adjust photomultiplier tube (PMT) voltages to maximize fluorescence resolution and dynamic range, using methods like the stain index voltration [80].
- Controls: Always include positive and negative controls. Fluorescence Minus One (FMO) controls are essential for accurate gating in multicolor panels, as they provide the most accurate assessment of background and spillover spreading [81] [80]. The use of isotype controls alone is not considered sufficient [81].
- Antibody Titration: Titrate all fluorescent reagents to determine the concentration that provides the optimal stain index. Using antibodies at saturating but not excessive concentrations maximizes sensitivity and minimizes background [81].
- Spillover Compensation: For conventional flow cytometers, spectral spillover between channels must be compensated using single-stain controls. This arithmetic subtraction is critical for accurate multi-parameter data [81].
- Gating Strategy: Define and report a clear gating strategy that includes a viability dye to exclude dead cells, as light scatter properties (FSC/SSC) alone are not sufficient for determining viability [80].
Achieving high reproducibility in flow cytometry across multiple laboratories demands a disciplined approach to standardization. This involves meticulous panel design, adherence to optimized staining protocols that minimize cell loss and preserve epitopes, rigorous instrument calibration, and the consistent use of appropriate controls. By implementing the detailed protocols and quality control measures outlined in this document, researchers and drug development professionals can significantly enhance the reliability and comparability of their data on intracellular stem cell markers, thereby accelerating scientific discovery and therapeutic development.
Conclusion
Mastering the flow cytometry analysis of intracellular stem cell markers requires a holistic approach that integrates robust foundational knowledge, a meticulously optimized staining protocol, proactive troubleshooting, and rigorous analytical validation. The methodologies outlined provide a framework for generating high-quality, reproducible data essential for accurately defining the pluripotent state of stem cells. As the field advances, the adoption of standardized protocols and validation practices will be paramount. Future developments in high-parameter spectral cytometry, novel fluorophores, and automated analysis hold the promise of deeper insights into stem cell heterogeneity, ultimately accelerating progress in regenerative medicine, drug discovery, and our fundamental understanding of developmental biology.
References
- 1. NaNog: A pluripotency homeobox (master) molecule - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4625207/]
- 2. Functional roles of pluripotency transcription factors in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3495805/]
- 3. safeguards pluripotency and mediates germline... | Nature Nanog [https://www.nature.com/articles/nature06403?error=cookies_not_supported&code=dca9bf71-f1ef-4bdb-b966-17b2a6d2a917]
- 4. Nanog and transcriptional networks in embryonic stem cell ... [https://www.nature.com/articles/7310125]
- 5. , as a Nanog cancer key in tumor progression stem cell marker [https://pubmed.ncbi.nlm.nih.gov/35337852/]
- 6. The dynamical organization of the core pluripotency ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2023.1125015/full]
- 7. Intracellular Flow Cytometry | Intracellular Staining [https://www.bdbiosciences.com/en-us/learn/applications/multicolor-flow-cytometry/intracellular-flow-cytometry]
- 8. 5 Steps to Intracellular Flow Cytometry [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometry-assays-reagents/intracellular-flow-cytometry.html]
- 9. Flow Cytometry Protocol for Cell Surface Markers [https://www.rndsystems.com/resources/protocols/flow-cytometry-protocol-staining-membrane-associated-proteins-suspended-cells]
- 10. Flow Cytometry: a versatile tool for stem cell research [https://www.sciencedirect.com/science/article/abs/pii/S0006291X25017498]
- 11. Flow Cytometry Protocol | Abcam [https://www.abcam.com/en-us/technical-resources/protocols/flow-cytometry-for-intracellular-and-extracellular-targets]
- 12. Intracellular Flow Cytometry Staining Protocol [https://www.ptglab.com/news/blog/intracellular-flow-cytometry-staining-protocol/?srsltid=AfmBOopHvKuZpNMjxqoB8455cNVLtEm6CLuYxmu7mzPlGKfrNHtOn_dH]
- 13. Optimized, Efficient Measurement of the Expression ... [https://pubmed.ncbi.nlm.nih.gov/40028708/]
- 14. A fixation-compatible protocol for intracellular and surface ... [https://www.nature.com/articles/s41598-025-23698-1]
- 15. 7 Advanced Flow Cytometry Data Analysis Tips For Multi ... [https://expertcytometry.com/flow-cytometry-data-analysis-tips-for-multi-color-experiments/]
- 16. Reliable Protocols for Flow Cytometry Analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6727984/]
- 17. Flow Cytometry, Methanol Permeabilization Protocol [https://www.cellsignal.com/learn-and-support/protocols/protocol-flow-methanol-permeabilization?srsltid=AfmBOoqIE3ehtoObptSFps7jaHPwLNPOLgDVsPl5pC0DWyK_YPTjE9mE]
- 18. Publishing flow cytometry data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2822558/]
- 19. Combined Flow Cytometric Analysis of Surface and ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0068519]
- 20. Data analysis in flow cytometry [https://www.abcam.com/en-us/technical-resources/guides/flow-cytometry-guide/data-analysis-in-flow-cytometry]
- 21. Quick Tips on Flow Cytometry Data Analysis [https://www.bdbiosciences.com/en-us/learn/science-thought-leadership/blogs/flow-cytometry-data-analysis-quick-tips]
- 22. A Protocol for Culture and Characterization of Human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10506163/]
- 23. High-throughput robotic isolation of human iPS cell clones ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04414-2]
- 24. BestProtocols: Cell Preparation for Flow Cytometry Protocols [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/cell-preparation-flow-cytometery.html]
- 25. Introducing the Dish Soap Protocol: A Unified Approach for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12447862/]
- 26. Staining Intracellular Antigens for Flow Cytometry [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/staining-intracellular-antigens-flow-cytometry.html]
- 27. Intracellular Flow Cytometry [https://www.ptglab.com/products/flow-cytometry-antibodies/intracellular-flow-cytometry/?srsltid=AfmBOoqcN3xch8v9C8-ZUCXz9-wLixQ8ibOlhnrbpoz_3UOWq7E4uoHQ]
- 28. Flow Cytometry, Methanol Permeabilization Protocol [https://www.cellsignal.com/learn-and-support/protocols/protocol-flow-methanol-permeabilization?srsltid=AfmBOoo_x2ChXCDKI4JyRxaC8sHm_MHoqtcrdgVwo0P0wIfsZBXJ2BqF]
- 29. (IC) Intracellular Staining Flow Cytometry Protocol Using ... [https://www.rndsystems.com/resources/protocols/flow-cytometry-protocol-staining-intracellular-molecules-using-detergents]
- 30. Optimization of the Blocking and Signal Preservation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12462741/]
- 31. Optimizing Flow Cytometry Experiments-Part 2 How To Block ... [https://expertcytometry.com/optimizing-flow-cytometry-experiments-part2-how-to-block-samples-sample-blocking/]
- 32. Recommended controls for flow cytometry [https://www.abcam.com/en-us/technical-resources/guides/flow-cytometry-guide/recommended-controls-for-flow-cytometry]
- 33. Intracellular Flow Cytometry Staining Protocol [https://www.ptglab.com/news/blog/intracellular-flow-cytometry-staining-protocol/?srsltid=AfmBOoqmKEuiGP4fdWHL5P6AM-J7GIqwusADUTtUQIYwopuo2Xo3-izi]
- 34. Staining for Flow Cytometry [https://www.southernbiotech.com/staining-for-flow-cytometry/]
- 35. Newsletter: Cellular Markers for Flow ... - FluoroFinder Cytometry [https://fluorofinder.com/newsletter-cellular-markers-for-flow-cytometry-analysis/]
- 36. Designing a multicolour flow protocol | Abcam cytometry [https://www.abcam.com/en-us/technical-resources/guides/flow-cytometry-guide/designing-a-multicolor-protocol]
- 37. and Dyes | Proteintech Group Flow Cytometry Fluorophores [https://www.ptglab.com/support/flow-cytometry-protocol/flow-cytometry-fluorophores-and-dyes/]
- 38. Fluorochrome Faceoff Series: Performance Data ... [https://www.bdbiosciences.com/en-us/learn/science-thought-leadership/blogs/flow-cytometry-fluorochrome-performance-data-results]
- 39. Flow Cytometry Controls [https://www.fortislife.com/resources/antibody-resources/flow-cytometry-controls]
- 40. Flow Cytometry: An Overview - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5939936/]
- 41. Fc Blocking - McGovern Medical School - UTHealth Houston [https://med.uth.edu/imm/flow-cytometry-services/protocols-and-guidelines/fc-blocking/]
- 42. The Power of Reagent Titration in Flow Cytometry - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11506663/]
- 43. A gentle fixation and permeabilization method for ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/1709845/]
- 44. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOoqhyghaDjsd1rLE5EwL5bPlGHgDA44XFwNl-EvHJclbOSzUF34L]
- 45. Flow Cytometry Troubleshooting [https://www.antibodies.com/applications/flow-cytometry/flow-cytometry-troubleshooting]
- 46. 3 Requirements For Accurate Flow Cytometry Compensation [https://expertcytometry.com/requirements-for-accurate-flow-cytometry-compensation/]
- 47. Experimental Controls in Flow Cytometry: Optimization Guide [https://www.bosterbio.com/protocol-and-troubleshooting/flow-cytometry-optimization/experimental-controls?srsltid=AfmBOooyOCwG1PCqYXt5cb1OvNV41g6qtgNGq4vxqsDsSq7DHhrWuWuw]
- 48. BestProtocols: Viability Staining Protocol for Flow Cytometry [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/viability-staining-flow-cytometry.html]
- 49. Viability Dyes for Flow Cytometry: It's Not Just a Matter of ... [https://bitesizebio.com/22353/viability-dyes-for-flow-cytometry-its-not-just-a-matter-of-life-and-death/]
- 50. Mastering Multi-Color Panel Design in Flow Cytometry [https://www.elabscience.com/resources/flow-cytometry/1882?srsltid=AfmBOoqUyxh3NK7NmB6F-mkdE2ed2pjdakPOP1fkPzR_6k1_81jQhN5b]
- 51. EuroFlow standardization of flow cytometer instrument ... [https://www.nature.com/articles/leu2012122]
- 52. Dye-mediated Binding | McGovern Medical School [https://med.uth.edu/imm/flow-cytometry-services/protocols-and-guidelines/dye-mediated-binding/]
- 53. Optimisation of blocking during flow cytometry [https://www.listonlab.uk/blog/2025/9/25/optimisation-of-blocking-during-flow-cytometry.html]
- 54. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOoouIsaw4hV4f4PoRuDaU6wLqjN_9EtFQeNYtHEbrjVn89_1PQyN]
- 55. Spectral Flow Cytometry Panel Controls and Sample ... [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometry-learning-center/flow-cytometry-resource-library/flow-cytometry-methods/spectral-flow-cytometry-panel-controls-sample-preparation.html]
- 56. Flow Cytometry Protocols for Surface and Intracellular Antigen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4396953/]
- 57. A Survey of Flow Cytometry Data Analysis Methods - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2798157/]
- 58. Sample Preparation Tips for Accurate Flow Cytometry ... [https://www.labx.com/resources/sample-preparation-tips-for-accurate-flow-cytometry-results/5521]
- 59. LSR Unclogging Procedure - Flow Cytometry [https://flowcytometry.medicine.uiowa.edu/resources/lsr-unclogging-procedure]
- 60. Flow cytometry: rules for analyzers - Yale Research [https://research.yale.edu/flow-cytometry-rules-analyzers]
- 61. Assay Validation - Cytometry [https://cytometry.mlsascp.com/assay-validation.html]
- 62. Qualification of a flow cytometry-based method for the ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10830575/]
- 63. Calculating Inter- and Intra-Assay Coefficients of Variability [https://salimetrics.com/calculating-inter-and-intra-assay-coefficients-of-variability/]
- 64. Protocol Resources for Flow Cytometry [https://www.stemcell.com/technical-resources/methods-library/characterization-assays/protocols/flow-cytometry.html]
- 65. Validation of High-sensitivity Flow Cytometry for Reliable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10345173/]
- 66. Surface Marker Analysis by Flow Cytometry [https://www.thermofisher.com/us/en/home/references/protocols/neurobiology/neurobiology-protocols/surface-marker-analysis-by-flow-cytometry.html]
- 67. The Power of Flow Cytometry in MRD Detection in Oncology [https://www.cerbaresearch.com/article/blog-post-finding-a-needle-in-a-haystack-the-power-of-flow-cytometry-in-mrd-detection-in-oncology/]
- 68. Flow cytometry-based validation of soluble biomarker detection [https://pmc.ncbi.nlm.nih.gov/articles/PMC12648499/]
- 69. Impact of high-sensitivity flow cytometry on peri-transplant ... [https://www.nature.com/articles/s41598-025-91936-7]
- 70. Measurable Residual Disease Analysis by Flow Cytometry [https://www.mdpi.com/2072-6694/17/7/1155]
- 71. Intracellular Flow Cytometry Staining Protocol [https://www.ptglab.com/news/blog/intracellular-flow-cytometry-staining-protocol/?srsltid=AfmBOoob8CiuHe0FMDGzFiutWXaAMsoFpCN_3zCX1E41ekhYsVGkn22G]
- 72. Ten Tips for Successful Flow Cytometry [https://www.biotechniques.com/cell-and-tissue-biology/how-to-10-tips-for-successful-flow-cytometry/]
- 73. . Conventional Flow Cytometry VS Spectral Flow Cytometry [https://www.bio-techne.com/resources/blogs/conventional-flow-cytometry-vs-spectral-flow-cytometry]
- 74. Antibodies 101: Conventional vs Spectral Flow Cytometry [https://blog.addgene.org/antibodies-101-conventional-vs-spectral-flow-cytometry]
- 75. Spectral Flow Cytometry Fundamentals [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometry-learning-center/flow-cytometry-resource-library/flow-cytometry-methods/spectral-flow-cytometry-fundamentals.html]
- 76. Designing a High-Parameter Spectral Panel for Immune ... [https://www.bdbiosciences.com/en-us/learn/science-thought-leadership/blogs/designing-high-parameter-spectral-panel]
- 77. Flow cytometry markers guide [https://www.abcam.com/en-us/knowledge-center/flow-cytometry/flow-cytometry-markers]
- 78. . Spectral : Key... | KCAS Bio vs Conventional Flow Cytometry [https://kcasbio.com/blogs/unveiling-the-future-of-flow-cytometry-conventional-vs-spectral-flow/]
- 79. Protocol for immunocytochemistry of stem cells [https://www.reprocell.com/blog/protocol-for-immunocytochemistry-of-stem-cells]
- 80. Improving the Quality and Reproducibility of Flow Cytometry in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6670040/]
- 81. Principles of Advanced Flow Cytometry: A Practical Guide [https://pmc.ncbi.nlm.nih.gov/articles/PMC10802916/]