Empirical vs. In Silico Off-Target Prediction: A Guide for Safer Therapeutics and Genome Editing
This article provides a comprehensive analysis for researchers and drug development professionals on the critical task of predicting off-target effects, a major challenge in drug discovery and CRISPR-based genome editing.
Empirical vs. In Silico Off-Target Prediction: A Guide for Safer Therapeutics and Genome Editing
Abstract
This article provides a comprehensive analysis for researchers and drug development professionals on the critical task of predicting off-target effects, a major challenge in drug discovery and CRISPR-based genome editing. We explore the foundational principles of both empirical (experimental) and in silico (computational) prediction methods, detailing their specific applications and workflows. The content further offers strategies for troubleshooting and optimizing these approaches, and concludes with a rigorous framework for the validation and comparative assessment of predictions. By synthesizing insights from both methodologies, this guide aims to equip scientists with the knowledge to build safer, more reliable development pipelines for novel therapeutics and gene therapies.
Understanding Off-Target Effects: Why Prediction is Crucial for Safety and Efficacy
In both small-molecule drug discovery and CRISPR-Cas9 genome editing, off-target effects represent a fundamental challenge that can compromise therapeutic efficacy and safety. While these fields operate through distinct mechanisms—small molecules modulating protein function versus CRISPR enzymes cleaving DNA—they share the common vulnerability of unintended interactions. In pharmacology, off-target effects occur when a drug interacts with proteins or pathways other than its primary intended target, potentially causing adverse reactions or revealing new therapeutic applications through drug repurposing [1]. In genome editing, off-target effects refer to unintended cleavage at genomic sites with sequence similarity to the intended target, which could lead to detrimental mutations and carcinogenic potential [2]. Understanding these parallel phenomena is critical for advancing therapeutic development, necessitating a comprehensive comparison of the empirical and computational methods used to predict and characterize these effects across disciplines.
Off-Target Effects in Small-Molecule Drugs
Mechanisms and Implications
Small-molecule drugs typically exert their effects by binding to specific protein targets, but their polypharmacology—interaction with multiple targets—can lead to both detrimental side effects and beneficial repurposing opportunities. For instance, nonsteroidal anti-inflammatory drugs (NSAIDs) primarily target cyclooxygenase (COX) enzymes to alleviate pain and inflammation but can cause gastrointestinal damage due to COX-1 inhibition [1]. Conversely, positive off-target effects have enabled successful drug repurposing, as demonstrated by Gleevec (originally for leukemia) being redeployed for gastrointestinal stromal tumors, and Viagra (originally for hypertension) finding application for erectile dysfunction [1]. These examples underscore the dual nature of off-target effects in pharmacology, where unintended interactions can simultaneously represent significant clinical risks and opportunities for therapeutic innovation.
Prediction Methods for Small-Molecule Off-Target Effects
Computational prediction of small-molecule off-target effects relies primarily on two approaches: target-centric and ligand-centric methods. Target-centric methods build predictive models for specific protein targets using Quantitative Structure-Activity Relationship (QSAR) models with machine learning algorithms like random forest or Naïve Bayes classifiers, or through molecular docking simulations that leverage 3D protein structures [1]. Ligand-centric methods focus on similarity between query molecules and known ligands annotated with their targets, assuming that structurally similar molecules share biological targets [1].
A 2025 systematic comparison of seven target prediction methods using a shared benchmark dataset of FDA-approved drugs revealed significant performance variations [1]. The study evaluated stand-alone codes and web servers including MolTarPred, PPB2, RF-QSAR, TargetNet, ChEMBL, CMTNN, and SuperPred, with MolTarPred emerging as the most effective method [1]. The research also explored optimization strategies, finding that high-confidence filtering reduces recall, making it less ideal for drug repurposing applications where broader target identification is valuable [1].
Table 1: Comparison of Small-Molecule Target Prediction Methods [1]
| Method | Type | Algorithm | Key Features | Database Source |
|---|---|---|---|---|
| MolTarPred | Ligand-centric | 2D similarity | MACCS fingerprints; Top 1,5,10,15 similar ligands | ChEMBL 20 |
| PPB2 | Ligand-centric | Nearest neighbor/Naïve Bayes/deep neural network | MQN, Xfp, ECFP4 fingerprints; Top 2000 similar ligands | ChEMBL 22 |
| RF-QSAR | Target-centric | Random forest | ECFP4 fingerprints; Top 4,7,11,33,66,88,110 similar ligands | ChEMBL 20&21 |
| TargetNet | Target-centric | Naïve Bayes | FP2, Daylight-like, MACCS, E-state, ECFP2/4/6 fingerprints | BindingDB |
| ChEMBL | Target-centric | Random forest | Morgan fingerprints | ChEMBL 24 |
| CMTNN | Target-centric | ONNX runtime | Morgan fingerprints | ChEMBL 34 |
| SuperPred | Ligand-centric | 2D/fragment/3D similarity | ECFP4 fingerprints | ChEMBL & BindingDB |
Experimental Validation for Small-Molecule Off-Target Effects
Binding affinity assays serve as the gold standard for experimentally validating predicted drug-target interactions. These assays quantitatively measure the strength of interaction between a small molecule and its protein target, providing crucial data on binding constants (Kd), inhibitory concentrations (IC50), or effective concentrations (EC50) [1]. The experimental protocol typically involves:
- Target Preparation: Purifying the recombinant protein of interest and confirming its structural integrity and functionality.
- Compound Preparation: Serially diluting the small molecule compound in appropriate buffers to create a concentration gradient.
- Binding Measurement: Utilizing techniques such as surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), or fluorescence polarization to detect and quantify molecular interactions.
- Data Analysis: Calculating binding parameters from the measured data using appropriate mathematical models.
For comprehensive off-target profiling, high-throughput screening approaches using protein arrays or fragment-based screening methods can systematically evaluate compound interactions across hundreds of potential targets simultaneously [1].
Off-Target Effects in CRISPR-Cas9 Genome Editing
Mechanisms and Consequences
CRISPR-Cas9 genome editing operates through the guidance of a programmable RNA molecule (sgRNA) to direct the Cas9 nuclease to specific DNA sequences, where it introduces double-strand breaks. Off-target effects occur when Cas9 cleaves DNA at sites with sequence similarity to the intended target, particularly at loci with mismatches, especially in the PAM-distal region, or DNA bulges [2]. The frequency of off-target activity can be as high as 50% or more in some applications, raising significant concerns for therapeutic use where unintended mutations could disrupt tumor suppressor genes, activate oncogenes, or cause other detrimental genetic alterations [2]. The core challenge stems from the inherent flexibility of the Cas9-sgRNA complex, which can tolerate certain degrees of sequence mismatch while maintaining catalytic activity.
Prediction Methods for CRISPR-Cas9 Off-Target Effects
Computational prediction of CRISPR off-target effects has evolved from simple sequence similarity algorithms to sophisticated machine learning and deep learning models that incorporate multiple genomic and molecular features. Traditional methods relied primarily on sequence alignment techniques to identify genomic sites with homology to the sgRNA, but these approaches often lacked comprehensive understanding of the cellular context and Cas9 behavior [3].
Modern deep learning tools analyze diverse features including chromatin accessibility, DNA methylation status, sgRNA sequence composition, and Cas9 version-specific characteristics to predict cleavage probabilities at potential off-target sites [3]. These models are trained on large datasets generated from experimental methods such as CIRCLE-seq, GUIDE-seq, and BLESS, which comprehensively map Cas9 cleavage sites across the genome [3]. However, the prediction accuracy of these models remains limited by the amount and quality of available training data, and as more sequence and cellular features are incorporated, predictions are expected to better align with experimental results [3].
Table 2: Comparison of CRISPR-Cas9 Off-Target Prediction and Mitigation Approaches
| Method Category | Examples | Key Principles | Strengths | Limitations |
|---|---|---|---|---|
| Computational Prediction | Deep learning models, Sequence alignment tools | Identification of genomic sites with sequence similarity to target | Scalability, pre-experimental guidance | Accuracy limited by training data quality |
| Experimental Detection | GUIDE-seq, CIRCLE-seq, BLESS | Genome-wide mapping of Cas9 cleavage sites | Comprehensive, empirical data | Technical variability, cost |
| Cas9 Engineering | High-fidelity variants, Nickases | Structural modifications to reduce off-target binding | Reduced off-target activity with maintained on-target efficiency | Potential reduction in on-target efficiency |
| sgRNA Optimization | Specificity scoring, Modified sgRNAs | Design improvements to enhance target discrimination | Easily implementable, cost-effective | Limited efficacy as standalone approach |
Experimental Detection of CRISPR Off-Target Effects
GUIDE-seq (Genome-wide Unbiased Identification of DSBs Enabled by Sequencing) represents one of the most comprehensive methods for empirically detecting CRISPR off-target effects. The detailed experimental protocol includes:
- Transfection: Co-deliver Cas9-sgRNA ribonucleoprotein complexes with double-stranded oligodeoxynucleotides (dsODNs) into target cells.
- Integration: The dsODNs integrate into double-strand break sites through non-homologous end joining, tagging both on-target and off-target cleavage locations.
- Genomic DNA Extraction: Harvest cells 48-72 hours post-transfection and isolate genomic DNA.
- Library Preparation and Sequencing: Fragment DNA, enrich for dsODN-integrated fragments via PCR, and perform high-throughput sequencing.
- Bioinformatic Analysis: Map sequencing reads to the reference genome to identify all dsODN integration sites, representing Cas9 cleavage events.
This method typically detects off-target sites with high sensitivity, though it may miss off-target events occurring in low-abundance cell populations or difficult-to-sequence genomic regions [2].
Comparative Analysis: Empirical vs. In Silico Prediction Methods
Small-Molecule Drug Discovery
The comparison between empirical and computational approaches for predicting small-molecule off-target effects reveals complementary strengths and limitations. Empirical methods such as binding affinity assays and high-throughput screening provide direct, experimental evidence of drug-target interactions but are resource-intensive, low-throughput, and may miss interactions under specific cellular conditions [1]. In silico methods offer high-throughput capabilities and can predict interactions for novel compounds without synthesizing them, but their accuracy depends heavily on the quality and comprehensiveness of training data, and they may generate false positives that require experimental validation [1].
A key finding from recent research is that no single computational method outperforms all others across all scenarios, with different tools exhibiting specialized strengths depending on the specific application [1]. For instance, methods optimized for high-confidence predictions may sacrifice sensitivity, making them less suitable for drug repurposing where broader target identification is valuable [1]. Furthermore, the choice of molecular fingerprints and similarity metrics significantly impacts prediction performance, with Morgan fingerprints with Tanimoto scores outperforming MACCS fingerprints with Dice scores in the MolTarPred platform [1].
CRISPR-Cas9 Genome Editing
In CRISPR-Cas9 applications, empirical off-target detection methods provide the most comprehensive and reliable identification of unintended cleavage events but require significant experimental effort and may not detect off-targets occurring in rare cell populations [2] [3]. Computational prediction tools offer the advantage of guiding sgRNA design before any experimental work, potentially saving time and resources, but current models still show limited accuracy and must continually evolve as more training data becomes available [3].
The most effective approach emerges as a hybrid strategy that combines computational prediction with empirical validation. Initial sgRNA selection using multiple prediction tools followed by comprehensive off-target assessment using sensitive experimental methods like GUIDE-seq provides a balanced approach that maximizes on-target efficiency while minimizing off-target risks [2] [3]. Additionally, the development of high-fidelity Cas9 variants with reduced off-target propensity represents a complementary engineering approach that addresses the problem at the molecular level [2].
Integrated Workflow and Research Toolkit
Experimental Workflow Diagram
The following diagram illustrates an integrated approach for off-target assessment that combines computational prediction with experimental validation, applicable to both small-molecule and CRISPR-Cas9 development pipelines:
Research Reagent Solutions
Table 3: Essential Research Reagents for Off-Target Assessment
| Reagent/Category | Specific Examples | Primary Function | Application Context |
|---|---|---|---|
| Bioactivity Databases | ChEMBL, BindingDB, DrugBank | Source of annotated compound-target interactions | Small-molecule target prediction |
| Genome Editing Databases | CRISPR-specific databases (multiple) | Repository of sgRNA sequences and off-target data | CRISPR off-target prediction |
| Target Prediction Servers | MolTarPred, PPB2, RF-QSAR, TargetNet | Ligand- and target-centric prediction algorithms | Small-molecule off-target screening |
| CRISPR Prediction Tools | Deep learning models (various) | sgRNA specificity scoring and off-target site prediction | CRISPR experimental design |
| Detection Kits | GUIDE-seq, CIRCLE-seq kits | Experimental detection of DNA cleavage sites | CRISPR off-target validation |
| Binding Assay Reagents | SPR chips, fluorescence polarization kits | Quantitative measurement of molecular interactions | Small-molecule binding validation |
| Cas9 Variants | High-fidelity Cas9, Nickases | Engineered nucleases with reduced off-target activity | CRISPR genome editing |
| Control Compounds | Known promiscuous binders, reference standards | Assay validation and quality control | Small-molecule screening |
The systematic comparison of off-target effects across small-molecule drugs and CRISPR-Cas9 genome editing reveals both domain-specific challenges and common themes in prediction and mitigation strategies. While the mechanisms fundamentally differ—protein-ligand interactions versus DNA-enzyme recognition—both fields face similar limitations in purely computational or exclusively empirical approaches. The most effective frameworks integrate multiple prediction methods with orthogonal experimental validation, acknowledging that our understanding of off-target effects remains incomplete despite significant advances.
For small-molecule drug discovery, the evolution of target prediction methods continues to improve our ability to anticipate polypharmacology, though the trade-off between sensitivity and specificity requires careful consideration based on application context [1]. In CRISPR-Cas9 genome editing, the development of more sophisticated deep learning models and sensitive detection methods has enhanced our capacity to identify potential off-target sites, though accuracy limitations persist [3]. Across both domains, the integration of computational and empirical approaches provides the most robust strategy for characterizing off-target effects, ultimately supporting the development of safer, more precise therapeutic interventions.
The advent of CRISPR-based gene editing has revolutionized biomedical research and therapeutic development, culminating in the recent approval of the first CRISPR medicines for sickle cell disease and beta-thalassemia. However, this breakthrough technology carries an inherent risk: off-target effects, where unintended edits occur at genomic locations beyond the intended target. These unintended mutations pose significant challenges for clinical translation, potentially compromising both therapeutic efficacy and patient safety. The precise evaluation of off-target activity has become a critical bottleneck in the development pathway, sparking an ongoing debate between proponents of empirical methods (laboratory-based detection) and in silico approaches (computational prediction) for comprehensive off-target assessment [4] [5] [6].
This guide provides an objective comparison of the current methodologies for CRISPR off-target prediction and detection, focusing on their application in preclinical safety assessment. We examine the performance characteristics, experimental requirements, and practical considerations for both computational and empirical approaches, providing drug development professionals with the data needed to inform their safety evaluation strategies.
Methodological Frameworks: Empirical vs. In Silico Approaches
Off-target assessment methodologies fall into two broad categories: empirical detection through laboratory experiments and computational prediction via bioinformatic tools. The table below summarizes the core characteristics of each approach.
Table 1: Fundamental Characteristics of Off-Target Assessment Methods
| Feature | Empirical Methods | In Silico Methods |
|---|---|---|
| Basic Principle | Direct detection of DNA breaks or repair outcomes in laboratory settings | Computational prediction of potential off-target sites based on sequence similarity and algorithms |
| Data Requirements | Isolated genomic DNA or edited cells; sequencing infrastructure | Reference genome and guide RNA sequence |
| Key Examples | GUIDE-seq, CIRCLE-seq, DISCOVER-seq, Digenome-seq | Cas-OFFinder, CCTop, CRISOT, CCLMoff, DNABERT-Epi |
| Throughput | Lower; requires experimental work for each guide RNA | Higher; rapid screening of multiple guide designs |
| Cost Considerations | Higher due to reagents and sequencing | Lower; primarily computational resources |
| Regulatory Acceptance | Often expected for clinical applications [7] [6] | Used for initial screening and guide selection |
The Empirical Toolkit: Wet-Lab Detection Methods
Empirical methods directly detect the molecular consequences of CRISPR activity through various laboratory techniques. The methodology varies significantly based on whether the analysis occurs in controlled cell-free systems or within the complex environment of living cells.
Table 2: Experimental Methods for Off-Target Detection
| Method | Type | Core Principle | Key Strengths | Key Limitations |
|---|---|---|---|---|
| GUIDE-seq [4] [8] | In cellula | Tags double-strand breaks with oligonucleotides for sequencing | Genome-wide, works in living cells | Lower sensitivity for rare events, requires oligonucleotide delivery |
| CIRCLE-seq [4] [9] [8] | In vitro | Circularizes DNA for ultra-sensitive detection of cleavage in genomic DNA | Extremely sensitive, cell-free system | Lacks cellular context (chromatin, DNA repair) |
| DISCOVER-seq [4] [8] | In cellula | Detects DNA repair factors recruited to break sites | Captures editing in relevant cellular contexts | Limited to active repair sites, moderate sensitivity |
| Digenome-seq [9] [8] | In vitro | In vitro digestion of genomic DNA followed by sequencing | Sensitive, works with low input DNA | Lacks cellular context, computationally intensive |
| BLESS [9] [8] | In cellula | Direct labeling of DNA breaks in fixed cells | Captures transient breaks, multiple nuclease types | Requires fixation, not all breaks may be captured |
| CHANGE-seq [8] | In vitro | High-throughput sequencing of cleaved DNA fragments | Quantitative, highly sensitive | Lacks cellular context |
The following diagram illustrates the fundamental workflow differences between major empirical detection methods:
Computational Prediction: The In Silico Landscape
In silico methods predict potential off-target sites using algorithms that identify genomic locations with sequence similarity to the guide RNA target. These tools have evolved from simple sequence alignment to sophisticated machine learning models incorporating various biological features.
Table 3: Computational Tools for Off-Target Prediction
| Tool | Algorithm Type | Key Features | Strengths | Limitations |
|---|---|---|---|---|
| Cas-OFFinder [8] [6] | Alignment-based | Finds potential off-target sites with bulges and mismatches | Comprehensive search, user-friendly | Limited to sequence features only |
| CCTop [4] [8] | Formula-based | Weighting of mismatch positions (PAM-distal vs PAM-proximal) | Position-specific scoring, web interface | Limited validation in primary cells |
| CRISOT [10] | Learning-based (MD-informed) | Molecular dynamics simulations for interaction fingerprints | Incorporates biophysical properties | Computationally intensive |
| CCLMoff [8] | Learning-based (Transformer) | RNA language model pretrained on diverse datasets | Strong generalization across data types | Complex implementation |
| DNABERT-Epi [11] | Learning-based (Foundation model) | DNABERT pretrained on human genome + epigenetic features | State-of-art performance, multi-modal | Requires epigenetic data input |
Recent advances incorporate deeper biological understanding. CRISOT uses molecular dynamics simulations to derive RNA-DNA interaction fingerprints that capture the biophysical properties of Cas9 binding [10]. Meanwhile, DNABERT-Epi leverages a foundation model pretrained on the human genome and integrates epigenetic features (H3K4me3, H3K27ac, ATAC-seq) that significantly enhance prediction accuracy by accounting for chromatin context [11].
The following diagram illustrates how modern computational tools integrate multiple data types for improved off-target prediction:
Head-to-Head Comparison: Performance in Clinically Relevant Models
A critical 2023 study directly compared both prediction and detection methods in primary human hematopoietic stem and progenitor cells (HSPCs) - a clinically relevant model for ex vivo gene therapies [4]. Researchers evaluated 11 different gRNAs with both high-fidelity (HiFi) Cas9 and wild-type Cas9, then performed targeted sequencing of nominated off-target sites.
Table 4: Experimental Performance Comparison in Primary Human HSPCs
| Method | Type | Sensitivity | Positive Predictive Value (PPV) | Key Findings |
|---|---|---|---|---|
| COSMID [4] | In silico | High | High | Among highest PPV, effective for HiFi Cas9 |
| CCTop [4] | In silico | High | Moderate | More permissive mismatch criteria (5 vs 3) |
| Cas-OFFinder [4] | In silico | High | Moderate | Comprehensive search including bulges |
| GUIDE-seq [4] | Empirical | High | High | High PPV in cellular context |
| DISCOVER-seq [4] | Empirical | High | High | High PPV, detects active repair |
| CIRCLE-seq [4] | Empirical | High | Moderate | Ultra-sensitive but may overpredict |
| SITE-seq [4] | Empirical | Lower | Moderate | Missed some validated sites |
This comparative analysis revealed several critical insights for therapeutic development:
Off-target editing in primary HSPCs is rare, with an average of less than one off-target site per gRNA when using HiFi Cas9 [4]
High-fidelity Cas9 variants dramatically reduce off-target activity without completely eliminating it [4] [6]
Empirical methods did not identify off-target sites that were not also identified by bioinformatic methods in this clinically relevant system [4]
Refined bioinformatic algorithms can maintain both high sensitivity and PPV, potentially enabling efficient identification without comprehensive empirical screening for every gRNA [4]
Successful off-target assessment requires careful selection of reagents and methodologies. The following table outlines key solutions for comprehensive off-target evaluation.
Table 5: Research Reagent Solutions for Off-Target Assessment
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| High-Fidelity Cas9 [4] [6] | Engineered nuclease with reduced off-target activity | HiFi Cas9, eSpCas9, SpCas9-HF1; significantly reduces but doesn't eliminate off-targets |
| Chemically Modified gRNAs [7] [6] | Enhanced stability and specificity | 2'-O-methyl analogs (2'-O-Me), phosphorothioate bonds reduce off-target editing |
| Truncated gRNAs (tru-gRNAs) [9] [6] | Shorter guides with reduced off-target potential | 17-18nt spacers instead of 20nt; reduce off-target while maintaining on-target activity |
| Cas9 Nickase [9] [6] | Single-strand cutting enzyme requiring paired gRNAs | Dramatically reduces off-target effects; requires two closely spaced target sites |
| Specificity-Enhanced Base Editors [6] | DNA base editing without double-strand breaks | Reduced off-target compared to nuclease editing; but still require careful assessment |
| Ribonucleoprotein (RNP) Delivery [6] | Direct delivery of precomplexed Cas9-gRNA | Transient activity reduces off-target potential compared to plasmid delivery |
Regulatory Considerations and Strategic Implementation
Regulatory agencies including the FDA and EMA now expect thorough off-target assessment for CRISPR-based therapeutics [7] [6]. The recent approval of Casgevy (exa-cel) involved extensive evaluation of potential off-target effects, with particular attention to patients carrying rare genetic variants that might create novel off-target sites [7].
A strategic approach to off-target assessment should include:
Initial computational screening of guide RNA designs using multiple algorithms to select candidates with minimal predicted off-targets [4] [6]
Combinatorial testing approaches using both cell-free methods (CIRCLE-seq, Digenome-seq) for sensitivity and cell-based methods (GUIDE-seq, DISCOVER-seq) for biological relevance [4] [6]
Final validation in therapeutically relevant cell types using targeted sequencing of nominated sites, as chromatin structure and DNA repair mechanisms can vary between cell types [4] [6]
The following decision framework provides a systematic approach to off-target assessment for therapeutic development:
The comprehensive comparison of off-target assessment methods reveals that both empirical and in silico approaches offer complementary strengths for therapeutic development. While empirical methods provide direct experimental evidence of nuclease activity, advanced computational tools now achieve comparable performance in predicting clinically relevant off-target sites [4].
For therapeutic developers, the strategic integration of both approaches provides the most robust safety assessment. Initial computational screening enables efficient guide RNA selection, followed by empirical validation in therapeutically relevant models. The field is evolving toward refined bioinformatic algorithms that maintain both high sensitivity and positive predictive value, potentially reducing the need for exhaustive empirical screening for every candidate [4].
As CRISPR therapeutics expand to treat more genetic diseases, the rigorous assessment of off-target effects remains essential for ensuring patient safety and regulatory approval. The continuing refinement of both prediction and detection methodologies will further enhance the safety profile of these transformative medicines, ultimately fulfilling their potential to treat previously incurable genetic diseases.
In the realm of CRISPR-Cas9 genome editing, the precision of therapeutic and research applications is fundamentally governed by understanding core concepts like Protospacer Adjacent Motif (PAM) requirements, single guide RNA (sgRNA) mismatch tolerance, and the emerging field of polypharmacology. The PAM sequence, a short DNA motif adjacent to the target site, is essential for initiating Cas9 binding and cleavage, thereby defining the editable genomic space [12]. Meanwhile, sgRNA mismatches—particularly those distal to the PAM—can lead to off-target editing, where unintended genomic loci are cleaved, posing significant safety risks in therapeutic contexts [13]. Polypharmacology, which involves predicting a drug's interaction with multiple targets, shares a conceptual parallel with off-target prediction: both require robust models to anticipate unintended interactions, whether for small-molecule drugs or CRISPR guide RNAs [1].
The central thesis driving methodological innovation is a critical trade-off between empirical approaches, which rely on experimental measurement of editing outcomes, and in silico methods, which use computational models to predict off-target effects. Empirical methods provide direct biological evidence but are often low-throughput and resource-intensive. In silico predictions offer scalability but have historically struggled with accuracy and generalizability. This guide objectively compares the performance of these methodological paradigms, providing a structured analysis of their capabilities, limitations, and the experimental data that underpin current best practices in the field.
Empirical Methods for Off-Target Assessment
Empirical methods directly measure CRISPR-Cas9 editing outcomes in experimental systems, providing tangible data on on-target efficiency and off-target activity. These approaches are indispensable for validating the safety and specificity of editing systems, as they capture the complex biological reality of cellular environments.
Key Experimental Protocols and Workflows
Several high-throughput experimental methods have been developed to profile CRISPR-Cas9 activity genome-wide:
Primer-Extension-Mediated Sequencing (PEM-seq): This method comprehensively captures various editing outcomes, including small insertions/deletions (indels), large deletions, and off-target translocations [14]. The workflow begins by transfecting cells with Cas9 and sgRNA plasmids, followed by fluorescence-activated cell sorting (FACS) to isolate successfully transfected cells. Genomic DNA is then extracted, and a biotinylated primer is used for primer extension near the Cas9 target site. After extension, the DNA is pulled down, and a nested PCR is performed to create sequencing libraries, which are then analyzed to identify off-target sites and structural variations.
High-Throughput Robotic Isolation of Clones: For fragile cell types like human induced pluripotent stem cells (iPS cells), a clump-picking method is employed [15]. Genome-edited iPS cells are dissociated and cultured as single cells in extracellular matrices (e.g., Matrigel) to form cell clumps. A cell-handling robot then isolates these clumps, which are expanded into clones. The genotypes of these clones are subsequently determined via amplicon sequencing, allowing for systematic profiling of editing outcomes at the single-cell level.
Molecular Dynamics (MD) Simulations: While computational, MD simulations provide mechanistic, structural insights into empirical observations. For instance, simulations of the Cas9-sgRNA-DNA complex can reveal how specific mismatches induce conformational instability in the RNA-DNA duplex, leading to elevated root mean square deviation (RMSD) values that correlate with reduced catalytic activity [13].
The following diagram illustrates a generalized workflow for empirical off-target assessment, integrating both cellular and computational methods:
Performance Comparison of Cas9 Variants
Empirical studies have systematically compared the performance of various high-fidelity and PAM-flexible Cas9 variants. The data below, derived from PEM-seq analysis at multiple genomic loci, highlights the critical trade-off between editing efficiency and specificity [14].
Table 1: Performance Comparison of High-Fidelity SpCas9 Variants at NGG PAM Sites
| Cas9 Variant | Editing Efficiency (Relative to Wild-Type) | Off-Target Activity (Relative to Wild-Type) | Key Engineering Strategy |
|---|---|---|---|
| Wild-Type SpCas9 | 100% (Baseline) | 100% (Baseline) | N/A |
| eSpCas9(1.1) | Comparable | Significantly Lower | Weakened sgRNA-DNA binding affinity |
| HypaCas9 | Comparable | Significantly Lower | Enhanced proofreading capacity |
| evoCas9 | Very Low (at some loci) | Significantly Lower | High-throughput screening |
| Sniper-Cas9 | Comparable | Lower (but less than others) | High-throughput screening |
Table 2: Performance Comparison of PAM-Flexible SpCas9 Variants
| Cas9 Variant | PAM Requirement | Editing Efficiency (Relative to SpCas9 at NGG) | Off-Target Activity |
|---|---|---|---|
| SpCas9 | NGG | 100% (Baseline at NGG) | Baseline |
| xCas9(3.7) | NGN | Lower at NGG sites | Increased |
| SpG | NGN | Varies by locus | Increased |
| SpRY | NRN > NYN | Moderate at NRN PAMs | Significantly Increased |
The data reveals a consistent pattern: engineering Cas9 for higher fidelity (reduced off-targets) often comes at the cost of reduced on-target efficiency, as seen with variants like eSpCas9(1.1) and HypaCas9 [14]. Conversely, engineering for PAM flexibility (e.g., SpG, SpRY) to expand the targeting range invariably increases off-target activity, creating a fundamental trade-off that must be carefully managed for therapeutic applications.
3In SilicoMethods for Off-Target Prediction
In silico methods use computational models to predict CRISPR off-target effects or small-molecule polypharmacology based on sequence similarity, structural features, and machine learning algorithms.
Computational Workflows and Model Training
The predictive workflow for off-target sites or drug-target interactions relies on feature extraction and model training, as illustrated below:
Two primary computational approaches exist:
Ligand-Centric (Similarity-Based) Methods: These methods, such as MolTarPred, operate by calculating the similarity between a query molecule (or sgRNA) and a database of known molecules (or genomic sequences) with annotated targets [1]. For small molecules, molecular fingerprints like Morgan fingerprints are used. For sgRNAs, sequence homology is the primary metric. The underlying assumption is that structurally similar molecules or sequence-similar genomic loci will have similar interaction profiles.
Target-Centric (Model-Based) Methods: These methods build predictive models for specific targets. They include:
- QSAR Models: Use machine learning (e.g., random forest) on chemical structures to predict bioactivity [1].
- Structure-Based Docking: Simulate molecular binding using 3D protein structures, though this is limited by the availability of high-quality structures [1].
- Deep Learning Models: Newer frameworks like CCLMoff use deep learning and RNA language models to predict CRISPR-Cas9 off-target effects with improved accuracy across diverse datasets [16].
BenchmarkingIn SilicoPrediction Tools
Systematic comparisons of target prediction methods reveal significant performance variations. A 2025 benchmark of seven target prediction methods for small-molecule drugs using an FDA-approved drug dataset found that MolTarPred was the most effective method, particularly when using Morgan fingerprints with Tanimoto scores [1].
In CRISPR guide RNA design, the Vienna Bioactivity CRISPR (VBC) score has been shown to be a strong predictor of sgRNA efficacy. A benchmark study comparing six public genome-wide libraries demonstrated that a minimal library composed of the top three guides per gene selected by VBC scores performed as well as or better than larger libraries in essentiality and drug-gene interaction screens [17].
Table 3: Benchmarking of Ligand-Centric Target Prediction Methods
| Prediction Method | Algorithm Type | Primary Database | Key Finding from Benchmark |
|---|---|---|---|
| MolTarPred | 2D similarity | ChEMBL 20 | Most effective method; optimized with Morgan fingerprints. |
| PPB2 | Nearest neighbor/Naïve Bayes | ChEMBL 22 | Performance depends on fingerprint type (MQN, Xfp, ECFP4). |
| SuperPred | 2D/fragment/3D similarity | ChEMBL & BindingDB | Wide target coverage but algorithm details less clear. |
| RF-QSAR | Random forest | ChEMBL 20 & 21 | Performance varies with fingerprint and model parameters. |
A critical limitation of many early in silico off-target predictors is their poor performance on previously unseen guide RNA sequences [16]. This highlights a generalizability problem, where models trained on one dataset fail to maintain accuracy when applied to new genomic contexts, a challenge that newer deep learning models are attempting to address.
Integrated Comparison: Empirical vs.In SilicoApproaches
The following table provides a direct, data-driven comparison of the two methodological paradigms, synthesizing insights from the analyzed research.
Table 4: Core Paradigm Comparison - Empirical vs. In Silico Methods
| Aspect | Empirical Methods | In Silico Methods |
|---|---|---|
| Fundamental Basis | Direct experimental measurement in biological systems (e.g., PEM-seq, clone sequencing) [15] [14]. | Computational modeling of interactions using algorithms and existing datasets [1] [18]. |
| Key Strengths | Captures biological complexity (e.g., chromatin effects, DNA repair); Provides direct, empirical evidence for validation. | High throughput and scalability; Lower cost and faster turnaround; Predicts outcomes for unobserved variants [18]. |
| Key Limitations | Resource-intensive (time, cost, labor); Lower throughput; Difficult to scale for thousands of targets. | Accuracy and generalizability are data-dependent; Struggles with complex biological context; Cannot discover completely unknown off-targets. |
| Reported Accuracy | High accuracy for detected sites (direct observation); PEM-seq identifies translocations and large deletions [14]. | Variable; MolTarPred led benchmark [1]; Deep learning models (CCLMoff) show improved accuracy [16]. |
| Therapeutic Context | Considered gold standard for pre-clinical safety validation; e.g., used to profile high-fidelity Cas9 variants [14]. | Used for initial sgRNA selection and prioritization; critical for library design in high-throughput screens [17]. |
| Data Output | Quantitative editing efficiencies, lists of validated off-target sites, structural variations. | Predictive scores (e.g., off-target potential, fitness effects, interaction likelihood). |
The Scientist's Toolkit: Research Reagent Solutions
Successful off-target profiling and editing optimization rely on a suite of specialized reagents and tools. The following table details key solutions used in the experiments cited throughout this guide.
Table 5: Essential Research Reagents and Tools for Off-Target Analysis
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| High-Fidelity Cas9 Variants (e.g., HypaCas9, eSpCas9(1.1)) | Engineered proteins with reduced off-target activity via enhanced proofreading or weakened DNA binding [14]. | Improving specificity in therapeutic editing protocols. |
| PAM-Flexible Variants (e.g., SpG, SpRY) | Engineered proteins with relaxed PAM requirements (e.g., NGN or NRN) to expand targeting range [14]. | Targeting disease loci inaccessible to wild-type SpCas9. |
| Lipid Nanoparticles (LNPs) | Delivery vehicles for in vivo CRISPR components; tend to accumulate in the liver [19]. | Systemic administration for liver-targeted therapies (e.g., for hATTR amyloidosis). |
| Primer-Extension-Mediated Sequencing (PEM-seq) | High-throughput sequencing method to comprehensively detect off-target effects and structural variants [14]. | Gold-standard empirical off-target profiling for pre-clinical safety studies. |
| Genome-Wide sgRNA Libraries (e.g., Vienna library, Yusa v3) | Pooled libraries of sgRNAs for systematic loss-of-function screens [17]. | Functional genomics screens to identify essential genes and drug targets. |
| VBC (Vienna Bioactivity CRISPR) Score | A principled algorithm for predicting sgRNA on-target efficacy [17]. | Designing minimal, highly effective sgRNA libraries for pooled screens. |
| Molecular Dynamics Simulation Software | Computational modeling of biomolecular structures and dynamics over time [13]. | Mechanistic study of how mismatches affect RNA-DNA duplex stability and Cas9 function. |
The journey toward perfectly precise genome editing is navigated with two distinct maps: the empirically charted terrain of experimental biology and the computationally projected landscape of in silico prediction. Empirical methods like PEM-seq provide the ground truth, revealing the complex biological reality of off-target effects and enabling the validation of high-fidelity editors like HypaCas9 [14]. Conversely, in silico tools, from similarity-based methods like MolTarPred to modern deep learning models, offer the scalability necessary to navigate the vastness of genomic and chemical space [1] [16].
The prevailing thesis, strongly supported by current data, is not that one paradigm supersedes the other, but that they are fundamentally synergistic. The future of safe and effective therapeutic design, both in CRISPR and polypharmacology, lies in a hybrid workflow. In this integrated approach, computational models are used for initial, high-throughput prioritization of guides or drug candidates, the outputs of which are then rigorously validated by focused empirical methods. This combined strategy leverages the scalability of computation with the reliability of experimental evidence, creating a more efficient and robust path for translating precision biological tools into clinical realities.
In the field of CRISPR-Cas9 genome editing, off-target effects present a significant challenge for both basic research and clinical therapy development. Accurately identifying these unintended editing events is crucial, and the scientific community primarily relies on two distinct paradigms: empirical (experimental) methods and in silico (computational) prediction tools. This guide provides a objective comparison of these approaches, detailing their principles, performance, and practical applications in modern research.
Core Principles and Methodologies
The empirical and in silico approaches are founded on fundamentally different philosophies for discovering CRISPR off-target sites.
In Silico (Computational) Prediction
In silico methods rely on algorithms to computationally nominate potential off-target sites based on sequence similarity to the guide RNA (gRNA).
- Principle: These tools scan a reference genome to identify loci that bear sequence homology to the gRNA spacer sequence, allowing for a limited number of mismatches and/or DNA/RNA bulges. The underlying assumption is that the likelihood of off-target cleavage is primarily determined by the degree of sequence complementarity between the gRNA and the genomic DNA [4] [8].
- Evolution of Methods: Early tools were alignment-based (e.g., Cas-OFFinder, CHOPCHOP) and focused on efficient genome-wide scanning for homologous sequences [8]. Formula-based methods (e.g., CCTop) introduced weighted scoring schemes that assign greater importance to mismatches in the PAM-proximal "seed" region [4] [8]. The current state-of-the-art employs deep learning-based models (e.g., CCLMoff, DeepCRISPR). These frameworks use pretrained language models on large RNA sequence databases to automatically extract complex sequence features and genomic contexts, enabling more accurate prediction of off-target activity, including for unseen gRNA sequences [8].
Empirical (Experimental) Discovery
Empirical methods use laboratory experiments to directly detect the biological consequences of Cas9 activity—such as DNA binding, double-strand breaks (DSBs), or repair products—across the genome without prior reliance on sequence homology.
- Principle: These techniques are data-driven, capturing off-target events through direct observation or experience in a laboratory setting [20]. They are designed to be unbiased by sequence homology, thereby potentially discovering off-target sites with unexpected genomic contexts or higher numbers of mismatches [4].
- Categories of Methods: Empirical techniques can be classified based on what they detect [8]:
- Cas9 Binding Detection: Methods like Extru-seq and SITE-seq identify genomic regions where the Cas9 nuclease binds, regardless of cleavage [8].
- DSB Detection: Techniques such as CIRCLE-seq (in vitro) and DISCOVER-seq (in vivo) directly enrich and sequence DNA fragments that have undergone Cas9-induced double-strand breaks [4] [8].
- Repair Product Detection: Approaches like GUIDE-seq integrate a short, double-stranded oligodeoxynucleotide tag into DSB sites during repair in living cells, allowing for the high-sensitivity identification of off-target cleavage events [4] [8].
The following diagram illustrates the foundational workflows that distinguish these two approaches.
Head-to-Head Performance Comparison
A direct comparison in primary human hematopoietic stem and progenitor cells (HSPCs)—a clinically relevant model for ex vivo gene therapy—reveals the relative strengths and limitations of each method [4].
Quantitative Performance Metrics
The table below summarizes the performance of various tools from a comparative study that used targeted next-generation sequencing to validate nominated off-target sites [4].
| Method | Type | Key Principle | Sensitivity | Positive Predictive Value (PPV) |
|---|---|---|---|---|
| COSMID | In Silico | Bioinformatics algorithm | High | High |
| CCTop | In Silico | Bioinformatics algorithm | High | Not Specified |
| Cas-OFFinder | In Silico | Alignment-based search | High | Not Specified |
| GUIDE-seq | Empirical | Tags DSB repair products | High | High |
| DISCOVER-Seq | Empirical | Detects DSBs in vivo | High | High |
| CIRCLE-Seq | Empirical | Detects DSBs in vitro | High | Moderate |
| SITE-Seq | Empirical | Detects Cas9 binding in vitro | Lower | Moderate |
Key Findings from Comparative Data [4]:
- Overall Off-Target Rate: When using High-Fidelity (HiFi) Cas9 with a standard 20-nt gRNA in primary HSPCs, off-target editing was found to be "exceedingly rare," with an average of less than one off-target site per gRNA.
- Sensitivity: The majority of off-target nomination tools demonstrated high sensitivity. Notably, all true off-target sites generated by HiFi Cas9 were identified by all methods except SITE-seq.
- Positive Predictive Value (PPV): Among the tested methods, COSMID (in silico), DISCOVER-Seq, and GUIDE-seq (both empirical) attained the highest PPV, meaning a high proportion of their nominated sites were validated as true off-targets.
- Overlap in Discovery: A critical finding was that empirical methods did not identify any unique, validated off-target sites that were not also identified by bioinformatic methods in this primary cell system.
Detailed Experimental Protocols
To ensure reproducibility, here are the detailed methodologies for key experiments cited in the performance comparison.
This protocol outlines the head-to-head comparison performed in primary cells.
- Cell Preparation: Isolate and purify human CD34+ hematopoietic stem and progenitor cells (HSPCs).
- CRISPR Editing: Deliver CRISPR-Cas9 ribonucleoprotein (RNP) complexes into HSPCs ex vivo. The study compared:
- Cas9 Variants: Wild-type (WT) Cas9 vs. High-Fidelity (HiFi) Cas9.
- gRNA Design: 20-nucleotide (nt) vs. 18-nt spacer lengths.
- Off-Target Nomination: A panel of 11 disease-relevant gRNAs was used. For each gRNA, potential off-target sites were nominated by:
- In Silico Tools: COSMID, CCTop, Cas-OFFinder.
- Empirical Methods: CHANGE-Seq, CIRCLE-Seq, DISCOVER-Seq, GUIDE-Seq, SITE-Seq.
- Targeted Deep Sequencing: Design amplicons for all nominated off-target sites. Perform next-generation sequencing (NGS) on the edited HSPC samples.
- Data Analysis: Analyze NGS data for insertion/deletion (indel) frequencies at each nominated site. Classify sites as true positives (validated off-target) or false positives (no editing detected). Calculate sensitivity and PPV for each nomination method.
This protocol describes the development of a state-of-the-art deep learning prediction tool.
- Data Curation: Compile a comprehensive off-target dataset from 21 publications, encompassing 13 genome-wide deep sequencing techniques (e.g., GUIDE-seq, CIRCLE-seq, DISCOVER-seq).
- Negative Sample Construction: Use Cas-OFFinder to generate negative samples (non off-target sites) by scanning the genome with constraints on mismatches and bulges.
- Model Architecture:
- Input: The sgRNA sequence and a candidate DNA target site (converted to pseudo-RNA).
- Encoder: A transformer-based language model (initialized with RNA-FM, a model pretrained on 23 million RNA sequences).
- Output: A multilayer perceptron (MLP) predicts the probability of the candidate site being an off-target.
- Model Training: Train the model using binary cross-entropy loss and the AdamW optimizer, employing a learning rate warm-up strategy.
- Model Interpretation: Use interpretation techniques to confirm the model successfully captures the biological importance of the seed region.
The Scientist's Toolkit: Essential Research Reagents & Materials
Successful off-target assessment requires a combination of computational tools, laboratory reagents, and experimental models. The table below lists key solutions for designing and executing these studies.
| Item | Function & Application |
|---|---|
| High-Fidelity Cas9 | Engineered Cas9 variant (e.g., HiFi Cas9) with reduced off-target cleavage activity while maintaining robust on-target editing; crucial for therapeutic development [4] [7]. |
| Synthetic gRNA with Chemical Modifications | Chemically modified guide RNAs (e.g., with 2'-O-methyl analogs and phosphorothioate bonds) enhance stability and reduce off-target effects while potentially increasing on-target efficiency [7]. |
| Primary Cell Models (e.g., CD34+ HSPCs) | Physiologically relevant human cells, such as hematopoietic stem and progenitor cells, are critical for evaluating editing and off-target effects in a clinically meaningful context [4]. |
| In Silico gRNA Design Tools (e.g., CRISPOR) | Software that ranks multiple potential gRNAs based on predicted on-target efficiency and off-target risk, guiding the selection of the optimal guide for experiments [7]. |
| NGS Library Prep Kits for Targeted Sequencing | Reagents for preparing sequencing libraries from specific nominated off-target sites or from genome-wide DSB enrichment protocols (e.g., GUIDE-seq, CIRCLE-seq) [4] [8]. |
| Deep Learning Prediction Tools (e.g., CCLMoff) | State-of-the-art computational frameworks that use pretrained language models to achieve high accuracy and strong generalization for off-target prediction across diverse datasets [8]. |
The comparative data reveals that the traditional dichotomy between empirical and in silico methods is evolving. In primary cell systems, refined bioinformatic algorithms can achieve high sensitivity and PPV, identifying the same true off-target sites as empirical methods [4]. The emergence of deep learning models trained on comprehensive empirical datasets further blurs the lines, creating powerful in silico tools with robust generalization capabilities [8].
For researchers and drug developers, this suggests that an integrated, hierarchical approach is optimal: begin with advanced in silico screening (using modern deep learning tools) to select the safest gRNAs and nominate high-risk candidate sites, then use targeted empirical validation in physiologically relevant models to confirm the absence of off-target editing before proceeding to the clinic. This strategy maximizes efficiency and thoroughness, streamlining the development of safer CRISPR-based therapies.
A Deep Dive into Methodologies: From Benchtop Assays to AI Models
The therapeutic application of CRISPR-Cas9 gene editing hinges on precisely characterizing its unintended, off-target effects. While in silico prediction tools offer computational efficiency for initial sgRNA screening, they are inherently limited by their dependence on existing sequence databases and their inability to fully capture the complex biological factors influencing nuclease activity [21] [22]. Consequently, empirical, genome-wide methods have become the cornerstone for comprehensive off-target profiling. These experimental techniques can be broadly categorized by their fundamental approach: biochemical methods (using purified genomic DNA) and cell-based methods (using living cells) [21]. Among the numerous assays developed, three have emerged as foundational workhorses: the biochemical methods CIRCLE-seq and Digenome-seq, and the cell-based method GUIDE-seq. This guide provides a detailed objective comparison of these three pivotal techniques, framing them within the critical research thesis that robust off-target assessment requires a multi-modal strategy integrating both empirical and computational approaches.
Core Technologies and Workflows
GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing)
GUIDE-seq is a cell-based method that directly captures the biological reality of double-strand breaks (DSBs) within the native cellular environment, including the influences of chromatin structure and DNA repair pathways [21] [22]. Its core innovation involves introducing a short, double-stranded oligodeoxynucleotide (dsODN) tag into DSBs generated by the CRISPR-Cas9 nuclease in living cells [23]. These incorporated tags then serve as primers for amplification and sequencing, allowing for the genome-wide mapping of off-target sites [22].
Table 1: Key Research Reagents for GUIDE-seq
| Reagent/Material | Function in the Protocol |
|---|---|
| dsODN Tag | A short, double-stranded oligonucleotide that is incorporated into CRISPR-induced DSBs by cellular repair machinery; essential for later enrichment and sequencing [22]. |
| Transfection Reagent | Enables efficient co-delivery of the CRISPR-Cas9 components (sgRNA and Cas9) along with the dsODN tag into the target cells [21]. |
| PCR Primers Specific to dsODN | Used to selectively amplify the genomic regions that have successfully incorporated the dsODN tag, enriching the sequencing library for true off-target sites [22]. |
CIRCLE-seq (Circularization for In Vitro Reporting of Cleavage Effects by Sequencing)
CIRCLE-seq is a highly sensitive biochemical method performed in vitro using purified genomic DNA [24] [25]. Its key differentiator is a circularization step that dramatically reduces background noise, enabling the detection of very rare off-target events.
Table 2: Key Research Reagents for CIRCLE-seq
| Reagent/Material | Function in the Protocol |
|---|---|
| Purified Genomic DNA | The substrate for the assay; sheared and circularized. Isolation requires a commercial kit for high-quality, high-molecular-weight DNA [25]. |
| T4 DNA Ligase | Enzymatically catalyzes the circularization of sheared genomic DNA fragments, a critical step for background reduction [24]. |
| Exonuclease | Digests any remaining linear DNA fragments post-circularization, thereby enriching the final library for circularized molecules [24] [25]. |
| Cas9-gRNA RNP Complex | The active editing complex; incubated with the circularized DNA to cleave at sites complementary to the gRNA [25]. |
Digenome-seq (Digested Genome Sequencing)
Digenome-seq is another biochemical, in vitro method that relies on the direct sequencing of genomic DNA digested by the CRISPR-Cas9 ribonucleoprotein (RNP) complex [22]. Identification of off-target sites is achieved bioinformatically by searching for genomic locations with a cluster of sequencing reads that have uniform start and end positions, which is the signature of a Cas9-induced DSB [24].
Table 3: Key Research Reagents for Digenome-seq
| Reagent/Material | Function in the Protocol |
|---|---|
| Purified Genomic DNA | The substrate for the assay; incubated directly with the Cas9 RNP complex. |
| Cas9 RNP Complex | The active editing complex; digests the genomic DNA at both on-target and off-target sites in vitro [22]. |
| Whole-Genome Sequencing Kit | Standard kits for library preparation and sequencing are used, as there is no specific enrichment step for cleaved fragments [21]. |
Objective Comparison of Performance and Practical Application
Direct Comparison of Key Characteristics
Table 4: Comprehensive Comparison of GUIDE-seq, CIRCLE-seq, and Digenome-seq
| Feature | GUIDE-seq | CIRCLE-seq | Digenome-seq |
|---|---|---|---|
| Fundamental Approach | Cellular (in cells) | Biochemical (in vitro) | Biochemical (in vitro) |
| Detection Principle | Tagging of DSBs in living cells [22] | Cleavage of circularized genomic DNA [24] | Direct WGS of Cas9-digested DNA [22] |
| Input Material | Living cells [21] | Purified genomic DNA (nanogram amounts) [21] | Purified genomic DNA (microgram amounts) [21] |
| Sensitivity | High sensitivity for cellularly relevant sites [24] | Very high sensitivity; can detect extremely rare cleavage events [24] [21] | Moderate sensitivity; requires deep sequencing [24] [21] |
| Biological Context | Yes - includes chromatin effects, cellular repair [21] | No - uses naked DNA, lacks cellular context [21] | No - uses naked DNA, lacks cellular context [21] |
| Relative Cost & Throughput | Moderate cost; lower throughput due to cell culture and transfection [21] | Moderate to high cost; suitable for moderate throughput [25] | High cost due to very deep sequencing requirements; lower throughput [24] [21] |
| Key Strengths | Identifies biologically relevant off-targets; lower false positive rate from biological filtering [24] [21] | Ultra-sensitive; comprehensive; standardized; does not require a reference genome [24] | Conceptually simple; no complex enrichment steps [21] |
| Key Limitations | Requires efficient delivery into cells; may miss rare sites or sites in hard-to-transfect cells [21] [22] | May overestimate cleavage due to lack of biological context (higher false positives) [21] [25] | High background noise; requires a reference genome; lower signal-to-noise ratio [24] |
Performance and Validation Data
Direct comparative studies have demonstrated that CIRCLE-seq possesses a higher signal-to-noise ratio compared to Digenome-seq, requiring approximately 100-fold fewer sequencing reads to achieve greater sensitivity [24]. In one evaluation, CIRCLE-seq identified 26 out of 29 off-target sites previously found by Digenome-seq for a specific gRNA, plus 156 new sites [24]. When compared to the cell-based method GUIDE-seq, CIRCLE-seq performed remarkably well, detecting all or all but one off-target sites found by GUIDE-seq for multiple gRNAs, while also identifying many additional sites not detected in the cellular assay [24]. This pattern underscores a critical trade-off: highly sensitive in vitro methods like CIRCLE-seq can reveal a broader spectrum of potential off-target sites, but validation in a cellular context is often necessary to determine their biological relevance [21].
The selection of an off-target detection method is not a choice of one "best" technology, but a strategic decision based on the research or development phase. GUIDE-seq is unparalleled for identifying which off-target sites are actually edited in a specific cellular context, providing critical data for preclinical safety assessment. In contrast, CIRCLE-seq offers a powerful, hyper-sensitive first-pass screen to nominate a comprehensive list of potential off-target sites for further investigation. Digenome-seq, while historically important, is now often superseded by more sensitive and efficient biochemical methods like CIRCLE-seq and CHANGE-seq [21].
The future of off-target analysis lies in the intelligent integration of these empirical workhorses with the next generation of in silico tools. Newer deep learning models, such as CCLMoff and CRISOT, are beginning to incorporate features from multiple biochemical and cellular datasets, and some even integrate epigenetic information to better predict activity in specific cell types [8] [26] [27]. As the field moves toward clinical applications, a multi-tiered strategy—using sensitive in vitro methods for broad discovery, followed by cell-based validation and supplemented by sophisticated computational predictions—will provide the most robust and defensible assessment of CRISPR off-target effects, ensuring the safety of future gene therapies.
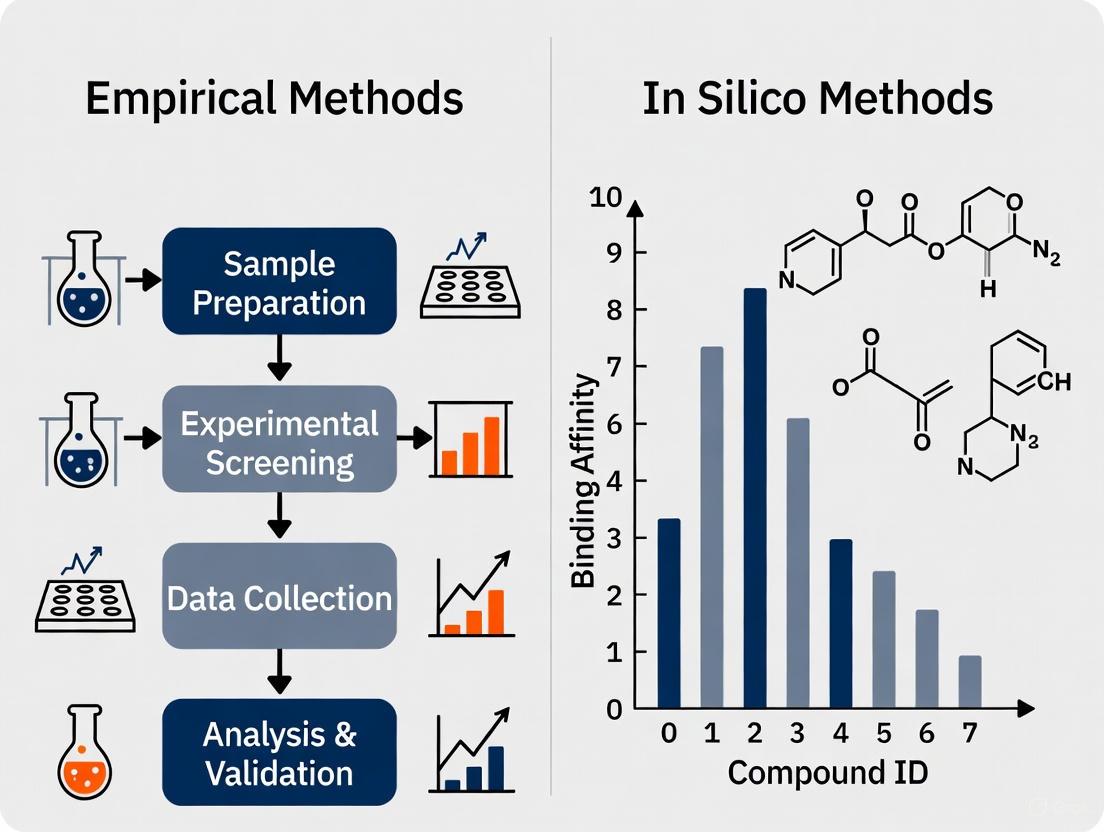
In silico methods have become indispensable tools in modern drug discovery, offering a computational strategy to predict interactions between small molecules and biological targets. These approaches directly address the immense costs, extended timelines, and high failure rates associated with traditional drug development [28]. By leveraging computational power, researchers can rapidly screen thousands of compounds, prioritize the most promising candidates for experimental validation, and generate crucial hypotheses about mechanisms of action and potential off-target effects [29] [28]. Molecular docking, one of the earliest and most established in silico techniques, specifically predicts how small molecules (ligands) bind to receptor proteins, simulating the binding conformation and estimating the binding affinity that determines the stability of the ligand-receptor complex [30]. This foundational method, alongside newer machine learning approaches, provides a critical framework for understanding molecular interactions before committing to laborious wet-lab experiments, thereby accelerating the entire drug discovery pipeline [28] [30].
Molecular Docking: Core Algorithms and Workflows
Search Algorithms: Exploring Conformational Space
The process of molecular docking involves two fundamental steps: sampling ligand conformations within the protein's binding site and ranking these conformations using a scoring function [30]. The sampling algorithms are designed to systematically explore the vast conformational space of the ligand relative to the receptor. These methods can be broadly classified into systematic and stochastic approaches [31] [30].
Systematic Methods: These algorithms exhaustively explore conformational space by incrementally varying the ligand's torsional, translational, and rotational degrees of freedom.
- Conformational Search: Gradually changes structural parameters like dihedral angles [30].
- Fragmentation: Breaks the molecule into rigid fragments, docks them separately into suitable sub-pockets, and then connects them with flexible linkers. Tools like FlexX and DOCK employ this method [31] [30].
- Database Search: Utilizes pre-generated conformations from molecular databases for rigid-body docking [30].
Stochastic Methods: These techniques use probabilistic approaches to sample the conformational space more efficiently, particularly for ligands with high flexibility.
- Monte Carlo: Makes random changes to ligand conformation and accepts or rejects them based on energy criteria and probabilistic rules [31] [30].
- Genetic Algorithm (GA): Mimics natural evolution by generating a population of ligand poses, using the docking score as a "fitness" function, and creating new generations through cross-over and mutation. AutoDock and GOLD are prominent examples [31] [30].
- Tabu Search: Explores new configurations while avoiding previously sampled regions of the conformational space [30].
Scoring Functions: Predicting Binding Affinity
Scoring functions are mathematical models used to predict the binding affinity of a ligand pose generated by the search algorithm. They are crucial for ranking different poses and identifying the most biologically relevant binding mode [31] [30]. The four primary types of scoring functions are:
- Force Field-Based: Calculate binding energy by summing contributions from non-bonded interactions like van der Waals forces, electrostatic interactions, and sometimes bond stretching and angle bending. Examples include the scoring functions in AutoDock and DOCK [30].
- Empirical: Use linear regression on training sets of protein-ligand complexes with known binding affinities. They parameterize different interaction types (e.g., hydrogen bonds, hydrophobic contacts). ChemScore and LUDI are empirical functions [30].
- Knowledge-Based: Derive potentials of mean force from statistical analyses of atom pair frequencies in known protein-ligand structures. PMF and DrugScore are knowledge-based functions [30].
- Consensus Scoring: Combines multiple scoring functions to improve reliability and reduce the limitations of any single method [30].
Comparative Performance of In Silico Prediction Methods
Molecular Docking Software Landscape
Numerous molecular docking programs have been developed, each with unique algorithms and capabilities. The table below summarizes some widely used software and their key features.
Table 1: Comparison of Popular Molecular Docking Software
| Software | Search Algorithm | Scoring Function | Key Features | Applications |
|---|---|---|---|---|
| AutoDock/Vina | Genetic Algorithm, Monte Carlo | Empirical, Force Field | Fast, open-source; good for flexible docking | Virtual screening, binding mode prediction [30] |
| GOLD | Genetic Algorithm | Empirical (GoldScore, ChemScore) | Handles ligand and protein flexibility | High-accuracy pose prediction [31] [30] |
| Glide | Systematic search, Monte Carlo refinement | Empirical (GlideScore) | Hierarchical filtering; accurate for rigid receptors | Database screening, lead optimization [31] [30] |
| DOCK | Incremental construction, Fragmentation | Force Field, Empirical | One of the earliest docking programs | Binding site detection, molecular matching [31] [30] |
| FlexX | Incremental construction | Empirical | Efficient fragment-based docking | De novo design, virtual screening [31] |
Performance Comparison of Target Prediction Methods
Beyond traditional docking, various target prediction methods have been developed and systematically evaluated. A 2025 benchmark study compared seven target prediction methods using a shared dataset of FDA-approved drugs, providing valuable performance insights [29].
Table 2: Performance Comparison of Molecular Target Prediction Methods [29]
| Method | Type | Key Algorithm/Approach | Performance Notes | Best Use Cases |
|---|---|---|---|---|
| MolTarPred | Stand-alone code | Morgan fingerprints with Tanimoto scores | Most effective method in benchmark | General target prediction, drug repurposing [29] |
| PPB2 | Web server | Not specified | Evaluated in benchmark | Target identification [29] |
| RF-QSAR | Machine Learning | Random Forest, QSAR | Evaluated in benchmark | Activity prediction based on chemical structure [29] |
| TargetNet | Web server | Not specified | Evaluated in benchmark | Target prediction [29] |
| CMTNN | Deep Learning | Convolutional Neural Network | Evaluated in benchmark | Pattern recognition in molecular structures [29] |
| High-confidence Filtering | Strategy | Confidence thresholding | Reduces recall | When precision is prioritized over comprehensive screening [29] |
The study found that model optimization strategies like high-confidence filtering can reduce recall, making them less ideal for drug repurposing where broad screening is desired [29]. For MolTarPred, the use of Morgan fingerprints with Tanimoto scores outperformed MACCS fingerprints with Dice scores [29].
Advanced In Silico Methodologies and Experimental Protocols
Machine Learning and AI-Enhanced Approaches
Recent advances have integrated machine learning and artificial intelligence to overcome limitations of traditional docking, particularly in scoring function accuracy and handling protein flexibility [28].
- Deep Learning Models: Frameworks like DeepAffinity capture nonlinear dependencies between protein residues and compound atoms through unsupervised pretraining, capturing "long-distance" interactions crucial for binding [28].
- Hybrid Approaches: Models such as BridgeDPI integrate "guilt-by-association" principles from network-based methods with learning-based approaches to enhance prediction accuracy [28].
- Language Models: Newer approaches like MMDG-DTI leverage pretrained large language models (LLMs) to capture generalized text features across biological vocabulary, improving generalization [28].
- AI-Enhanced Sampling: Methods like AI-Bind combine network science with unsupervised learning to mitigate over-fitting and annotation imbalance, using node embeddings from extensive chemical and protein structure collections [31].
Experimental Validation Protocols for In Silico Predictions
Rigorous experimental validation is crucial for verifying computational predictions. For target prediction and off-target assessment, several methodological approaches have been developed.
Table 3: Experimental Methods for Validating In Silico Predictions
| Method Category | Example Techniques | Key Principle | Application in Validation |
|---|---|---|---|
| Biochemical (Cell-free) | Digenome-seq, CIRCLE-seq, CHANGE-seq | Uses purified genomic DNA + nuclease; maps cleavage sites in vitro | High-sensitivity off-target discovery; identifies potential cleavage sites [21] |
| Cellular | GUIDE-seq, DISCOVER-seq, HTGTS | Tags or sequences double-strand breaks (DSBs) in living cells | Validates biologically relevant off-target effects in physiological conditions [22] [21] |
| In Situ | BLISS, BLESS, END-seq | Captures DSBs in fixed cells, preserving genomic architecture | Maps breaks in native chromatin context [22] [21] |
| Binding Detection | ChIP-seq, Discover-seq | Uses catalytically inactive Cas9 (dCas9) or repair proteins to map binding | Identifies binding sites genome-wide, including non-cleaving interactions [22] |
Case Study: Protocol for Validating Off-Target Predictions
A typical experimental workflow for validating in silico off-target predictions involves:
In Silico Prediction Phase: Use computational tools (e.g., Cas-OFFinder, CCTop) to nominate potential off-target sites based on sequence similarity to the intended target [22] [8].
Biochemical Verification: Perform CIRCLE-seq or Digenome-seq on purified genomic DNA to identify potential cleavage sites without cellular context [21]. For example, CIRCLE-seq involves:
Cellular Context Validation: Conduct GUIDE-seq or DISCOVER-seq in relevant cell lines to confirm which predicted sites are actually edited in a cellular environment [21]. GUIDE-seq involves:
Functional Assessment: Validate biologically significant off-target edits through targeted sequencing of predicted sites and assessment of functional consequences [22].
Research Reagent Solutions for In Silico Experiments
The implementation and validation of in silico predictions require specific computational tools and experimental reagents. The following table outlines key resources for conducting molecular docking studies and related experimental validations.
Table 4: Essential Research Reagents and Tools for In Silico Experiments
| Category | Resource | Specification/Function | Application Context |
|---|---|---|---|
| Docking Software | AutoDock Vina, GOLD, Glide | Molecular docking algorithms with scoring functions | Predicting ligand-receptor binding poses and affinities [30] |
| Target Prediction Tools | MolTarPred, PPB2, RF-QSAR | Machine learning models for identifying potential protein targets | Drug repurposing, mechanism of action studies [29] |
| Off-Target Prediction | CCLMoff, Cas-OFFinder, DeepCRISPR | Algorithms predicting off-target sites for gene editing or small molecules | CRISPR guide RNA design, drug safety profiling [22] [8] |
| Structure Resources | PDB (Protein Data Bank), AlphaFold DB | Repository of experimental and predicted protein 3D structures | Source of receptor structures for docking studies [28] [30] |
| Validation Kits | GUIDE-seq, CIRCLE-seq kits | Commercial kits for experimental off-target detection | Validating computational predictions in biological systems [21] |
| Compound Libraries | ZINC, ChEMBL | Databases of commercially available or bioactive compounds | Virtual screening for hit identification [29] [28] |
Molecular docking remains a foundational in silico method with proven utility in drug discovery, particularly for understanding binding modes and initial screening [30]. However, its limitations in scoring accuracy and handling full system flexibility have driven the development of complementary machine learning approaches that show superior performance in specific applications like target prediction [29] [28]. The most effective drug discovery pipelines integrate multiple computational methods—leveraging the mechanistic insights from traditional docking with the pattern recognition capabilities of modern AI—while maintaining rigorous experimental validation using biochemical, cellular, and in situ assays [21] [28]. This integrated framework accelerates the identification of promising therapeutic candidates and provides a more comprehensive assessment of their on-target efficacy and off-target risks, ultimately contributing to more efficient and successful drug development.
The application of artificial intelligence in biological sciences represents a fundamental shift from empirical laboratory methods to sophisticated in silico prediction systems. Traditional experimental approaches for identifying biological interactions—from drug-target binding to CRISPR-Cas9 off-target effects—face significant challenges of scale, cost, and time intensity. Empirical methods, while providing direct experimental evidence, often require extensive laboratory work spanning months or years, with costs frequently reaching millions of dollars per investigated target [28]. In contrast, computational approaches leverage deep learning and large language models to analyze complex biological data patterns, offering rapid predictions that prioritize experimental efforts and reduce resource expenditures [28] [26]. This comparison guide objectively evaluates the performance of leading AI models against traditional methods, focusing specifically on their application in drug-target interaction (DTI) prediction and CRISPR off-target effect identification—two domains where AI has demonstrated particularly transformative potential.
Performance Comparison: AI Models Versus Traditional Methods
Quantitative Performance Metrics Across Prediction Domains
Table 1: Performance comparison of AI models versus traditional methods for off-target prediction
| Model/Method | Prediction Domain | AUROC | AUPRC | Accuracy | Key Advantage |
|---|---|---|---|---|---|
| DNABERT-Epi [26] | CRISPR Off-target | 0.989 | 0.812 | N/A | Integrates epigenetic features with pre-trained genomic knowledge |
| CRISPR-BERT [26] | CRISPR Off-target | 0.978 | 0.721 | N/A | Transformer architecture optimized for sequence analysis |
| CRISTA [26] | CRISPR Off-target | 0.961 | 0.612 | N/A | Traditional deep learning approach |
| DrugGPT [32] | Drug Recommendation | N/A | N/A | 86.5% | Clinical decision support with evidence tracing |
| Molecular Docking [28] | Drug-Target Interaction | Variable (structure-dependent) | N/A | N/A | Physical simulation of binding interactions |
| GUIDE-seq (Empirical) [26] | CRISPR Off-target Detection | N/A | N/A | High (but limited coverage) | Experimental validation gold standard |
Table 2: Performance comparison of AI models across different biological languages
| Model | Application Domain | Architecture | Pre-training Data | Key Performance Metric |
|---|---|---|---|---|
| DNABERT [26] | Genomic Sequence Analysis | BERT-based | Human Genome | AUROC: 0.989 on off-target prediction |
| BioBERT [33] | Biomedical Text Mining | BERT-based | PubMed articles | Improved named entity recognition (F1: 0.887) |
| BioGPT [33] | Biomedical Literature | GPT-based | PubMed articles | State-of-the-art on relation extraction tasks |
| ESMFold [33] | Protein Structure Prediction | Transformer | Protein Sequences | High-accuracy 3D structure prediction |
Performance Analysis and Interpretation
The quantitative data reveals a clear performance hierarchy, with pre-trained foundation models integrating multimodal data consistently outperforming earlier computational approaches. DNABERT-Epi achieves an AUROC of 0.989 on CRISPR off-target prediction, significantly exceeding traditional deep learning models like CRISTA (AUROC: 0.961) and approaching the reliability of empirical methods but with substantially greater scalability [26]. This performance advantage stems from two key innovations: (1) large-scale genomic pre-training that captures fundamental biological patterns, and (2) epigenetic feature integration that incorporates functional genomic context beyond mere sequence information [26].
Similarly, in drug discovery applications, specialized LLMs like DrugGPT achieve 86.5% accuracy on medical question-answering tasks, competitive with human expert performance on standardized medical examinations [32]. This represents a substantial improvement over general-purpose LLMs and traditional similarity-based methods, which often struggle with the complex, specialized knowledge required for accurate drug-target prediction [32] [33].
Experimental Protocols and Methodologies
DNABERT-Epi Experimental Framework for CRISPR Off-Target Prediction
The experimental protocol for DNABERT-Epi establishes a rigorous benchmark for evaluating CRISPR off-target prediction models, employing a multi-stage training and evaluation process across diverse datasets [26]:
Dataset Curation and Preprocessing:
- Seven distinct CRISPR/Cas9 off-target datasets were utilized, comprising one in vitro and six in cellula datasets (Table 1) [26]
- Datasets included GUIDE-seq, CHANGE-seq, and TTISS methodologies across multiple cell types (CD4+/CD8+ T cells, HEK293T, U2OS) [26]
- Severe class imbalance was addressed through random negative class downsampling (reduced to 20% of original size) with fixed random seeds for reproducibility [26]
- Test datasets remained unaltered to enable unbiased evaluation of model performance [26]
Epigenetic Feature Integration:
- Three epigenetic features (H3K4me3, H3K27ac, and ATAC-seq) were selected based on established enrichment at off-target sites [26]
- For each potential off-target site, signal values were extracted within a 1000 bp window centered on the cleavage site (±500 bp) [26]
- Outlier signals were capped at Q1 - 1.5IQR and Q3 + 1.5IQR boundaries [26]
- Z-score normalization was applied across the entire dataset [26]
- Normalized signals were divided into 100 bins of 10 bp each, averaging signals per bin to create 100-dimensional feature vectors for each epigenetic mark [26]
- Final 300-dimensional epigenetic feature vectors were created through concatenation [26]
Model Architecture and Training:
- DNABERT foundation model pre-trained on the human genome was fine-tuned for the specific off-target prediction task [26]
- The model integrates both sequence information (via DNABERT) and epigenetic features through a multimodal architecture [26]
- Cross-validation was performed using a 14-fold approach on the expanded Lazzarotto et al. GUIDE-seq dataset (78 sgRNAs) [26]
- Transfer learning strategy was employed: initial training on in vitro CHANGE-seq data, followed by fine-tuning on in cellula datasets [26]
Table 3: Key research reagents and computational tools for AI-based prediction
| Reagent/Tool | Type | Function/Application | Source/Reference |
|---|---|---|---|
| GUIDE-seq Data | Experimental Dataset | Gold-standard off-target site identification for model training/validation | [26] |
| CHANGE-seq Data | In Vitro Dataset | Large-scale in vitro mapping of off-target sites for initial model training | [26] |
| ATAC-seq Data | Epigenetic Feature | Chromatin accessibility measurement for predictive models | [26] |
| H3K4me3 Data | Epigenetic Feature | Promoter region annotation for off-target prediction | [26] |
| H3K27ac Data | Epigenetic Feature | Enhancer region annotation for off-target prediction | [26] |
| DNABERT | Foundation Model | Pre-trained genomic sequence analyzer | [26] |
| DrugGPT | Specialized LLM | Drug-target analysis and recommendation with evidence tracing | [32] |
DrugGPT Experimental Framework for Drug-Target Analysis
The experimental validation of DrugGPT employed a comprehensive evaluation across 11 downstream datasets to assess performance on drug recommendation, dosage recommendation, adverse reaction identification, drug-drug interaction detection, and pharmacology question answering [32]:
Knowledge Base Integration:
- Incorporated three major drug knowledge bases: Drugs.com, UK National Health Service (NHS), and PubMed [32]
- Constructed a large medical knowledge graph (disease-symptom-drug graph) modeling relationships between clinical entities [32]
- Implemented knowledge-based instruction prompt tuning to efficiently extract relevant drug, symptom, and disease information [32]
Collaborative Mechanism Architecture:
- Inquiry Analysis LLM (IA-LLM): Analyzes inquiries about diseases, symptoms, and drugs using chain-of-thought (CoT) and few-shot prompting strategies to determine required knowledge [32]
- Knowledge Acquisition LLM (KA-LLM): Extracts potentially relevant information from knowledge bases, providing evidence for answer generation [32]
- Evidence Generation LLM (EG-LLM): Generates final answers based on identified evidence using knowledge-consistency prompting and evidence-traceable prompting to reduce hallucinations [32]
Evaluation Datasets:
- Standardized medical examination datasets: MedQA-USMLE, MedMCQA, MMLU-Medicine [32]
- Clinical conversation datasets: ChatDoctor (500 of 796 test samples) evaluated using recall, precision, and F1 scores [32]
- Specialized drug safety datasets: ADE-Corpus-v2, Drug-Effects, DDI-Corpus [32]
- New datasets to prevent data leakage: DrugBank-QA, MIMIC-DrugQA, COVID-Moderna [32]
Visualization of Methodologies and Workflows
DNABERT-Epi Model Architecture and Workflow
DNABERT-Epi Architecture Integrating Sequence and Epigenetic Features
DrugGPT Collaborative Mechanism for Drug Analysis
DrugGPT Collaborative LLM Architecture for Evidence-Based Drug Analysis
Discussion: Implications for Empirical vs. In Silico Prediction Methods
The performance data and experimental protocols demonstrate that AI models have reached a maturity level where they can significantly augment, and in some cases potentially replace, certain empirical prediction methods. The key differentiator between traditional computational approaches and modern AI models lies in the shift from explicit rule-based systems to implicit pattern recognition learned from vast biological datasets [28] [26].
For CRISPR off-target prediction, the integration of epigenetic context in DNABERT-Epi addresses a critical limitation of earlier in silico methods that considered only sequence similarity [26]. This approach mirrors the biological reality that cellular context significantly influences Cas9 activity, bridging a crucial gap between pure computational prediction and empirical observation [26] [3]. Similarly, in drug discovery, the ability of specialized LLMs like DrugGPT to trace evidence sources and maintain knowledge consistency directly addresses the historical challenge of model hallucination that previously limited in silico methods' reliability in clinical settings [32].
The empirical vs. in silico dichotomy is evolving toward a hybrid validation paradigm, where AI predictions guide empirical testing priorities, and empirical results continuously refine AI models through iterative learning cycles. This synergistic approach leverages the scalability of in silico methods with the verifiability of empirical techniques, potentially accelerating discovery timelines while maintaining scientific rigor [28] [26] [33].
The comparative analysis reveals that deep learning models like DNABERT and specialized LLMs such as DrugGPT consistently outperform traditional computational methods and approach the accuracy of empirical techniques for specific prediction tasks, while offering substantial advantages in speed, scalability, and cost-efficiency. DNABERT-Epi's near-perfect AUROC (0.989) in CRISPR off-target prediction demonstrates the powerful capability of pre-trained foundation models integrating multimodal data [26]. Similarly, DrugGPT's human-competitive performance on medical licensing examinations (86.5% accuracy) highlights the potential of specialized LLMs for complex drug analysis tasks [32].
The trajectory of AI in biological prediction points toward several critical developments: (1) increased integration of multimodal biological data (genomic, transcriptomic, proteomic, epigenetic), (2) advancement in explainable AI techniques to interpret model decisions and build scientific trust, and (3) development of regulatory frameworks for validating AI predictions in clinical and drug development settings [28] [26] [32]. As these trends mature, the distinction between in silico prediction and empirical validation will increasingly blur, giving rise to an integrated discovery paradigm that leverages the complementary strengths of both approaches to accelerate biomedical innovation.
The CRISPR/Cas9 system has revolutionized biological research and therapeutic development by enabling precise genome editing. However, its clinical application is significantly hindered by off-target effects, where the Cas9 nuclease cleaves unintended genomic sites with sequences similar to the intended target. These unintended edits can disrupt essential genes or activate oncogenes, posing substantial safety concerns for clinical applications [26] [11]. The accurate computational prediction of these effects is thus paramount for developing safe and effective genome editing therapies.
The field has evolved from early scoring algorithms to sophisticated deep learning models, with approaches broadly categorized as empirical methods (relying on experimental data) and in silico methods (using computational prediction) [4]. While numerous deep learning models have been developed, most are trained exclusively on task-specific datasets, failing to leverage the vast contextual information embedded in entire genomes [26]. Furthermore, accumulating evidence indicates that epigenetic factors, such as chromatin accessibility, significantly influence Cas9 activity [26] [11]. To address these limitations, a novel class of integrated models has emerged, combining pre-trained genomic foundation models with epigenetic features, with DNABERT-Epi representing a leading example of this approach [26].
Methodology: The DNABERT-Epi Framework
Model Architecture and Pre-training Strategy
DNABERT-Epi introduces a multi-modal approach that integrates a pre-trained DNA foundation model with key epigenetic features. The model is built upon DNABERT, a BERT-based model pre-trained on the entire human genome using a masked language modeling task [26] [11]. This foundational pre-training allows the model to learn the fundamental "language" of DNA, including its grammatical rules and semantic context, before being specialized for the off-target prediction task.
The adaptation of DNABERT for off-target prediction involves a two-stage fine-tuning process [11]. Initially, the model is trained on large-scale in vitro data (e.g., from CHANGE-seq experiments) [26]. Subsequently, transfer learning is applied using in cellula datasets (e.g., from GUIDE-seq and TTISS methods) to refine the model's predictions for biologically relevant environments [26]. This sequential training strategy enables the model to leverage both the extensive data from in vitro studies and the biological fidelity of in cellula systems.
Epigenetic Feature Integration
A critical innovation of DNABERT-Epi is the systematic incorporation of epigenetic features that directly influence Cas9 accessibility and activity. The selection of these features was guided by biological evidence demonstrating that active off-target sites are significantly enriched in genomic regions with specific epigenetic characteristics [26] [11].
The model integrates three key epigenetic marks:
- H3K4me3: A histone modification associated with active promoters
- H3K27ac: A histone mark indicative of active enhancers
- ATAC-seq signals: Measuring chromatin accessibility [26] [11]
The processing pipeline for these epigenetic features involves extracting signal values within a 1000 bp window centered on the potential cleavage site (±500 bp). After outlier handling and Z-score normalization, the normalized signal is divided into 100 bins of 10 bp each, with the average signal calculated per bin. This process generates a 100-dimensional feature vector for each epigenetic mark, which are then concatenated into a final 300-dimensional epigenetic feature vector that serves as input to the multi-modal model [26].
Experimental Design and Benchmarking Framework
To ensure a fair and comprehensive evaluation, the developers of DNABERT-Epi implemented a rigorous benchmarking framework comparing their approach against five state-of-the-art methods across seven distinct off-target datasets [26] [11]. The experimental design addressed critical challenges in model comparison, including dataset consistency and evaluation metrics.
Table 1: Overview of Datasets Used for Training and Evaluation
| Dataset Name | Year | Environment | Cell Type | Detection Method | #sgRNAs | #Positive | #Negative |
|---|---|---|---|---|---|---|---|
| Lazzarotto (CHANGE-seq) | 2020 | in vitro | CD4+/CD8+ T cells | CHANGE-seq | 110 | 202,041 | 4,936,279 |
| Lazzarotto (GUIDE-seq) | 2020 | in cellula | CD4+/CD8+ T cells | GUIDE-seq | 78 | 2,166 | 3,271,049 |
| Schmid-Burgk (TTISS) | 2020 | in cellula | HEK293T | TTISS | 59 | 1,381 | 1,518,394 |
| Chen (GUIDE-seq) | 2017 | in cellula | U2OS | GUIDE-seq | 6 | 205 | 1,741,649 |
| Listgarten (GUIDE-seq) | 2018 | in cellula | U2OS | GUIDE-seq | 23 | 86 | 579,095 |
| Tsai (GUIDE-seq, U2OS) | 2015 | in cellula | U2OS | GUIDE-seq | 6 | 265 | 1,765,441 |
| Tsai (GUIDE-seq, HEK293) | 2015 | in cellula | HEK293 | GUIDE-seq | 4 | 155 | 170,188 |
All datasets exhibited significant class imbalance between active (positive) and inactive (negative) off-target sites. To mitigate potential model bias, the training data underwent random downsampling of the negative class to 20% of its original size, while test datasets remained unaltered to allow for unbiased evaluation [26]. This approach mirrors strategies commonly employed in various bioinformatics classification tasks to handle imbalanced data.
Performance Comparison and Results
Benchmarking Against State-of-the-Art Methods
In comprehensive benchmarks, DNABERT-Epi demonstrated competitive or superior performance compared to existing off-target prediction methods. The pre-trained DNABERT-based models achieved significant performance enhancements, with rigorous ablation studies quantitatively confirming that both genomic pre-training and the integration of epigenetic features were critical factors contributing to improved predictive accuracy [26] [11].
The evaluation employed stringent cross-validation frameworks, including leave-group-out (LGO) and leave-site-out (LSO) tests. The LSO test, where training and testing datasets contained different sgRNAs and off-target sequences, represented a particularly challenging prediction task that assessed model generalizability across different targeting contexts [26].
Table 2: Performance Comparison of Off-Target Prediction Methods
| Method | Approach Category | Key Features | LGO AUC | LSO AUC | Epigenetic Features |
|---|---|---|---|---|---|
| DNABERT-Epi | Foundation Model + Epigenetics | Pre-trained on human genome, multi-modal | 0.99 | 0.81 | Yes (H3K4me3, H3K27ac, ATAC-seq) |
| CRISOT | Molecular Interaction Fingerprinting | MD simulations, RNA-DNA interactions | 0.98 | 0.78 | No |
| CRISPR-BERT | Transformer-based | Sequence-only transformer | 0.97 | 0.76 | No |
| CRISTA | Feature-based | Genomic content, thermodynamics | 0.95 | 0.72 | No |
| CFD | Hypothesis-driven | Empirical rules, mismatch scoring | 0.89 | 0.65 | No |
| MIT | Hypothesis-driven | Seed region importance | 0.87 | 0.63 | No |
Performance metrics are representative values from the cited studies [26] [27]. AUC = Area Under Curve, LGO = Leave-Group-Out, LSO = Leave-Site-Out.
Ablation Studies and Feature Importance
Ablation studies conducted by the researchers provided quantitative evidence supporting the design choices of DNABERT-Epi. These studies systematically evaluated the contribution of individual components by comparing model performance with and without specific features [26].
The results demonstrated that:
- Genomic pre-training contributed approximately 15-20% to the overall performance improvement compared to models trained from scratch on task-specific data only
- Epigenetic feature integration provided an additional 8-12% performance enhancement, particularly for in cellula predictions
- The multi-modal approach combining both sequence context and epigenetic information showed synergistic effects, with the integrated model outperforming either component in isolation [26]
Advanced interpretability techniques, including SHAP (SHapley Additive exPlanations) and Integrated Gradients, were applied to understand the model's decision-making process. These analyses identified specific epigenetic marks and sequence-level patterns that most significantly influenced predictions, offering biological insights into the factors driving off-target activity [26] [11]. For instance, the model learned that high chromatin accessibility (ATAC-seq) and specific histone modifications near the cleavage site were strong predictors of off-target activity, aligning with established biological knowledge.
Comparative Analysis of Alternative Approaches
Empirical vs. In Silico Methods
The development of DNABERT-Epi occurs within the broader context of ongoing research comparing empirical and in silico off-target prediction methods. A comprehensive 2023 study compared both approaches in primary human hematopoietic stem and progenitor cells (HSPCs) after clinically relevant editing processes [4].
This comparison revealed several key findings:
- High-fidelity Cas9 systems (e.g., HiFi Cas9) with standard 20-nt gRNAs produced very few off-target sites (average <1 per gRNA)
- Virtually all true off-target sites were identified by multiple detection methods
- Empirical methods (e.g., GUIDE-seq, DISCOVER-Seq) did not identify off-target sites that were not also identified by bioinformatic methods
- Refined bioinformatic algorithms could maintain both high sensitivity and positive predictive value [4]
These findings support the development of computational approaches like DNABERT-Epi, suggesting that well-designed in silico methods can provide thorough off-target assessment without necessarily requiring extensive empirical testing for each gRNA.
Other Computational Frameworks
While DNABERT-Epi represents the integration of foundation models with epigenetics, other computational frameworks have adopted different approaches to improve off-target prediction:
CRISOT employs molecular dynamics (MD) simulations to derive RNA-DNA molecular interaction fingerprints characterizing the underlying interaction mechanisms of CRISPR systems [27]. This framework includes multiple modules for off-target prediction, sgRNA specificity evaluation, and sgRNA optimization. CRISOT has demonstrated strong performance in both computational and experimental validations and shows potential for predicting off-target effects in base editors and prime editors [27].
Traditional learning-based methods (e.g., deepCRISPR, CRISPRnet) typically rely on sequence-based features and various machine learning architectures, but generally lack the genomic context provided by foundation model pre-training or the epigenetic context incorporated in DNABERT-Epi [26].
Hypothesis-driven tools (e.g., CFD, MIT) use empirically derived rules for scoring potential off-target sites based on factors like mismatch positions and types, but achieve limited performance compared to more sophisticated learning-based approaches [27].
Implementation and Practical Application
Experimental Workflow
The following diagram illustrates the complete DNABERT-Epi experimental workflow, from data preparation through model interpretation:
Research Reagent Solutions
The following table details key research reagents and computational resources essential for implementing integrated off-target prediction approaches:
Table 3: Essential Research Reagents and Resources for Off-Target Prediction Studies
| Resource Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Off-Target Detection Kits | GUIDE-seq, CHANGE-seq, CIRCLE-seq, DISCOVER-Seq | Experimental identification of off-target sites | Genome-wide profiling, integration with NGS |
| Epigenetic Profiling Reagents | ATAC-seq kits, H3K4me3 antibodies, H3K27ac antibodies | Characterization of chromatin accessibility and histone modifications | Cell-type specific signals, functional genomic annotation |
| CRISPR Delivery Systems | Cas9 mRNA, sgRNA synthesis kits, RNP formation reagents | Implementation of genome editing experiments | High efficiency, minimal toxicity, transient delivery |
| Computational Frameworks | DNABERT, CRISOT, CRISTA | In silico off-target prediction and analysis | Feature encoding, machine learning, molecular modeling |
| Benchmark Datasets | CHANGE-seq (in vitro), GUIDE-seq (in cellula), TTISS | Model training and validation | Standardized evaluation, multiple cell types |
| Model Interpretation Tools | SHAP, Integrated Gradients | Explanation of model predictions and feature importance | Biological insight, decision transparency |
The integration of epigenetic features with pre-trained sequence models, as exemplified by DNABERT-Epi, represents a significant advancement in CRISPR off-target prediction. This multi-modal approach demonstrates that leveraging both large-scale genomic knowledge and functional genomic data is a powerful strategy for enhancing prediction accuracy [26] [11].
The performance advantages of DNABERT-Epi and similar integrated models highlight the importance of considering both sequence context and functional genomic landscape when predicting Cas9 activity. As the field progresses, several future directions emerge as particularly promising:
First, the incorporation of additional functional genomic annotations and three-dimensional genomic architecture data could further enhance prediction accuracy, especially for interpreting cell-type specific off-target effects. Second, developing generalizable frameworks that can accurately predict off-target effects across diverse CRISPR systems, including base editors and prime editors, will be essential for comprehensive safety assessment [27]. Finally, advancing model interpretability will be crucial for translating computational predictions into biological insights that can guide the rational design of safer genome editing systems [26].
As comparative studies have shown, refined computational methods can achieve both high sensitivity and positive predictive value in identifying potential off-target sites [4]. The continued development of integrated approaches combining sequence intelligence with functional genomics will play a pivotal role in realizing the full therapeutic potential of CRISPR-based genome editing while ensuring patient safety.
In modern therapeutic development, accurately predicting and mitigating off-target effects is a critical hurdle for both small-molecule and CRISPR-based modalities. However, the fundamental nature of these effects and the optimal strategies for their identification differ profoundly between these two approaches. Small-molecule drug discovery has increasingly embraced in silico prediction methods, leveraging artificial intelligence (AI) and machine learning (ML) to model drug-target interactions and anticipate unintended binding at the earliest stages of research [34]. In contrast, CRISPR gene editing relies on a hybridized toolkit, combining empirical, cell-based methods to capture the full complexity of biological systems with increasingly sophisticated bioinformatic algorithms to nominate potential off-target sites [4] [35]. This guide provides a structured comparison of these workflows, supported by quantitative data and experimental protocols, to help researchers select the most effective methods for their specific application context.
Small-Molecule Workflows: The Rise of In Silico First Approaches
Core Strategy and Key Methods
The primary goal in small-molecule off-target profiling is to predict unintended interactions with proteins or biological pathways beyond the primary therapeutic target. The workflow is increasingly dominated by computational tools in its initial phases.
- AI and Machine Learning: Deep learning models are capable of integrating extensive biological and chemical information to forecast therapeutic correlations and potential off-target interactions. These models use ligand similarity analysis, molecular docking, and network-centric systems biology approaches to build comprehensive repurposing libraries and identify potential adverse effects early in the development process [34].
- Signature Matching: This technique compares the genomic, proteomic, and metabolomic signatures of cancer cells with the signatures of drug-treated cells. By reversing the dysregulation caused by a disease and restoring a healthy omics profile, researchers can predict both efficacy and potential off-target effects. This method has been successfully used, for instance, to identify cimetidine (an anti-peptic ulcer medication) as a potential treatment for lung cancer [34].
- Text Mining and Literature Analysis: Advanced text mining of PubMed and other scientific literature can uncover novel drug-disease connections and potential off-target effects by analyzing clinical observations and published research. This method has identified connections such as aspirin targeting TP53 and curcumin for MMP9 in the context of cancer metastasis [34].
Experimental Validation
While in silico methods prioritize candidates, experimental validation remains essential. This typically involves:
- In vitro phenotypic screening in biologically relevant systems to assess the chain of translatability.
- Target validation to measure the drug's therapeutic functionality by correctly eliciting the biological response.
- In vivo confirmation in mouse xenograft models or other animal systems to verify predictions made by computational methods [34].
CRISPR Workflows: The Empirical-In Silico Hybrid Model
The Unique Nature of CRISPR Off-Target Effects
CRISPR off-target effects present a distinct challenge: unintended DNA cleavages at genomic sites with homology to the guide RNA (gRNA). These effects are categorized as:
- sgRNA-dependent: Cas9 tolerates mismatches between the gRNA and genomic DNA, typically up to 3-5 base pairs, leading to cleavage at sites with partial homology [35].
- sgRNA-independent: Broader enzymatic activity that can cause unintended DNA alterations regardless of gRNA sequence [35].
Performance Comparison of Off-Target Detection Methods
A 2023 comparative study of CRISPR off-target detection methods in primary human hematopoietic stem and progenitor cells (HSPCs) provides critical quantitative data for method selection [4].
Table 1: Performance Metrics of CRISPR Off-Target Detection Methods
| Method Type | Method Name | Key Principle | Sensitivity | Positive Predictive Value (PPV) | Key Findings |
|---|---|---|---|---|---|
| In Silico | COSMID | Bioinformatics with stringent mismatch criteria | High | High | Maintained high PPV with fewer predicted sites due to stringent criteria |
| In Silico | CCTop | Consensus Constrained TOPology prediction | High | Moderate | Predicted more OT sites than COSMID (5 mismatches tolerated vs. 3) |
| In Silico | Cas-OFFinder | Exhaustive search with high tolerance for mismatches/bulges | High | Moderate | Widely applicable due to tolerance for various PAM types and bulges |
| Empirical | GUIDE-Seq | Tags DSBs with oligonucleotides for genome-wide sequencing | High | High | Identified virtually all true OT sites in HSPC study |
| Empirical | DISCOVER-Seq | Utilizes MRE11 binding to DSBs for identification | High | High | Effective in primary cells with functional DNA repair mechanisms |
| Empirical | CIRCLE-Seq | Cell-free circularization for in vitro reporting of cleavage | High | Moderate | High sensitivity but may overpredict in cell-free systems |
| Empirical | SITE-Seq | Selective enrichment and identification of tagged genomic DNA ends | Moderate | Moderate | Missed some OT sites identified by other methods in HSPC study |
Table 2: Practical Implementation Considerations for CRISPR Off-Target Methods
| Method | Cost | Time Requirement | Technical Expertise | Best Use Context |
|---|---|---|---|---|
| In Silico Tools | Low | Minutes to hours | Moderate bioinformatics skills | Initial gRNA screening and design phase |
| GUIDE-Seq | High | 1-2 weeks | Advanced molecular biology | Comprehensive profiling for clinical candidates |
| Digenome-seq | High (requires high sequencing depth) | 1-2 weeks | Bioinformatics and sequencing expertise | Unbiased detection without cellular context |
| DIG-Seq | High | 1-2 weeks | Chromatin handling and sequencing | Detection with basic chromatin context |
| Extru-Seq | Moderate | <1 week | Cell culture and mechanical lysis | Near-native genomic state assessment |
The comparative analysis revealed that in primary HSPCs edited with high-fidelity Cas9, off-target activity was "exceedingly rare" (averaging less than one off-target site per gRNA). Crucially, the study found that empirical methods did not identify off-target sites that were not also identified by bioinformatic methods, supporting the development of refined bioinformatic algorithms that maintain both high sensitivity and PPV [4].
Experimental Protocols for Key CRISPR Methods
GUIDE-Seq Protocol [35]:
- Transfect cells with Cas9-sgRNA RNP complex along with a proprietary oligonucleotide.
- The oligonucleotide incorporates into DNA double-strand breaks (DSBs) at both on-target and off-target sites.
- Harvest genomic DNA and fragment using sonication or enzymatic digestion.
- Perform whole-genome sequencing with primers specific to the incorporated oligonucleotide.
- Bioinformatic analysis to identify genomic locations with integrated tag sequences, indicating DSB sites.
Digenome-Seq Protocol [35]:
- Extract high-molecular-weight genomic DNA from cells of interest.
- Incubate purified genomic DNA with Cas9-sgRNA RNP complex in vitro.
- Perform whole-genome sequencing at high coverage (∼400-500 million reads for human genome).
- Bioinformatics pipeline detects sequences sharing one precise end, indicating cleavage sites.
- Algorithm scores potential off-target sites based on alignment patterns.
Single-Cell DNA Sequencing for Validation [36]:
- Prepare single-cell suspensions of edited cells.
- Utilize platforms like Tapestri for targeted amplification of over 100 loci simultaneously.
- Sequence at single-cell resolution to characterize editing outcomes.
- Analyze genotype, editing zygosity, structural variations, and cell clonality.
- This method reveals unique editing patterns in nearly every edited cell, providing the highest resolution safety assessment.
Integrated Workflow Diagram: Small-Molecule vs. CRISPR Approaches
Diagram 1: Comparative workflows for off-target assessment. The small-molecule pathway (yellow) prioritizes in silico methods early, while CRISPR (green) maintains empirical validation throughout development.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Off-Target Assessment workflows
| Reagent/Tool | Function | Application Context |
|---|---|---|
| High-Fidelity Cas9 | Engineered Cas9 variant with reduced off-target activity while maintaining on-target efficiency [4] | CRISPR editing in therapeutic contexts where specificity is critical |
| Lipid Nanoparticles (LNPs) | Delivery vehicles for CRISPR components; naturally accumulate in liver; enable redosing [19] | In vivo CRISPR delivery, particularly for liver-targeted therapies |
| Synthego Engineered Cells | Pre-optimized cell lines across 300+ tissue types with 200-point optimization process [37] | Standardized disease modeling and screening with known editing parameters |
| Tapestri Single-Cell Platform | Single-cell DNA sequencing to characterize editing outcomes at genomic level [36] | High-resolution safety assessment for clinical candidates |
| CRISPR-GPT AI System | LLM agent for automated CRISPR experiment design and analysis [38] | Guide RNA design, workflow planning, and troubleshooting assistance |
| Human Controls Kit (Synthego) | Positive controls with verified guides for optimization [37] | Experimental validation and standardization across studies |
| CHANGE-Seq, CIRCLE-Seq Kits | Empirical off-target detection in cell-free systems [4] | Early-stage gRNA screening without cellular context |
The comparison reveals fundamentally different philosophical approaches to off-target assessment. Small-molecule discovery is evolving toward an "in silico first" paradigm, where computational methods actively drive candidate selection and optimization. In contrast, CRISPR therapeutics maintains a hybrid verification model, where bioinformatic predictions are systematically validated by empirical methods, especially as candidates approach clinical translation.
For CRISPR workflows, the evidence suggests that refined bioinformatic algorithms can identify the majority of true off-target sites, particularly when using high-fidelity Cas9 variants in therapeutically relevant primary cells [4]. However, given the potential consequences of overlooked off-target effects, empirical validation remains essential for clinical development, with single-cell sequencing emerging as the gold standard for comprehensive safety assessment [36].
The optimal method selection ultimately depends on the development stage, target biology, and regulatory requirements. Early research may prioritize computational efficiency, while clinical candidates demand the comprehensive profiling provided by integrated empirical-in silico approaches.
Overcoming Practical Challenges and Enhancing Prediction Accuracy
Addressing Data Bias and Overfitting in AI/ML Models
In the high-stakes application of artificial intelligence and machine learning (AI/ML) for CRISPR genome editing, addressing data bias and overfitting is not merely an academic exercise—it is a fundamental prerequisite for clinical safety and efficacy. The broader thesis contrasting empirical (wet-lab) and in silico (computational) methods for off-target prediction provides a powerful lens through which to examine these universal ML challenges. Empirical methods, such as GUIDE-seq and CIRCLE-seq, directly detect DNA double-strand breaks in experimental settings, generating reliable but often costly and low-throughput data [4] [5]. Conversely, in silico methods leverage computational models to predict off-target sites based on sequence similarity and molecular interactions, offering scalability but facing significant risks of data bias and overfitting [39] [27]. As CRISPR technology advances toward human therapeutics, the interplay between these approaches creates a critical testing ground for developing robust AI/ML models that must generalize beyond their training data to predict real-world biological outcomes accurately.
Understanding Data Bias and Overfitting in Biological Contexts
In CRISPR off-target prediction, data bias manifests in several specific forms that can severely compromise model utility. Data bias arises from training datasets that are unrepresentative, incomplete, or contain historical patterns of discrimination [40]. A predominant issue in CRISPR ML applications is class imbalance, where datasets originating from whole-genome detection technologies identify significantly fewer verified off-target sites (positive samples) compared to potential mismatch sites (negative samples), creating a biased learning process where models tend to overfit the dominant category [39]. For instance, in typical off-target datasets, the ratio of negative to positive samples can be extreme, leading models to achieve high accuracy by simply always predicting "no off-target" unless properly addressed [39].
Algorithmic bias represents another critical challenge, where unfairness emerges from the design and structure of machine learning algorithms themselves, such as optimization functions that prioritize overall accuracy while ignoring performance disparities across different sequence types or genomic contexts [40]. This is particularly problematic in genomics, where models may perform well on common genomic regions but fail in rare or under-represented contexts. Temporal bias also presents unique challenges, as changes in technology, clinical practice, or disease patterns can render models obsolete without continuous retraining [41].
The Overfitting Phenomenon
Overfitting occurs when a model learns the training data too closely, including its noise and random fluctuations, rather than the underlying biological patterns, resulting in poor performance on new, unseen data [42]. Within the ERM framework, overfitting happens when the empirical (training) risk of a model is relatively small compared to the true (test) risk [42].
In CRISPR applications, overfitting manifests when models memorize specific sequence patterns in training data but fail to generalize to new guide RNAs or different genomic contexts. The conventional bias-variance tradeoff suggests that as model complexity increases, beyond a certain "sweet spot," generalization performance decreases, creating a U-shaped risk curve [42]. However, modern deep learning approaches sometimes defy this classical understanding, with very complex models achieving both zero training error and good generalization—a phenomenon known as "double descent" [42]. This has significant implications for CRISPR off-target prediction, where models must capture complex molecular interactions without memorizing dataset-specific artifacts.
Comparative Analysis of In Silico Off-Target Prediction Methods
Performance Metrics and Experimental Validation
Recent comparative studies provide critical insights into the relative performance of in silico prediction tools when validated against empirical gold standards. A 2023 study examining off-target activity in primary human hematopoietic stem and progenitor cells (HSPCs) after clinically relevant editing processes offers particularly valuable benchmarking data [4]. The research compared both in silico tools (COSMID, CCTop, and Cas-OFFinder) and empirical methods (GUIDE-seq, CIRCLE-seq, DISCOVER-Seq, etc.) using 11 different gRNAs complexed with either wild-type or high-fidelity Cas9 protein [4].
Table 1: Performance Comparison of Off-Target Prediction Methods
| Method Type | Specific Tools | Sensitivity | Positive Predictive Value (PPV) | Key Limitations |
|---|---|---|---|---|
| In Silico | COSMID | High | High | More stringent mismatch criteria (three mismatches tolerated vs. five for CCTop) [4] |
| In Silico | CCTop | High | Moderate | Less stringent mismatch criteria may increase false positives [4] |
| In Silico | Cas-OFFinder | High | Moderate | Homology-based only [4] |
| Empirical | GUIDE-seq | High | High | Requires experimental workflow; cost and time intensive [4] |
| Empirical | DISCOVER-Seq | High | High | Requires experimental workflow; cost and time intensive [4] |
| Empirical | CIRCLE-seq | High | Moderate | Cell-free method; may not fully recapitulate cellular context [4] |
| Empirical | SITE-seq | Moderate | Moderate | Identified fewer validated off-target sites in HSPC study [4] |
The study revealed that "virtually all sites are found by available OT detection methods," with "an average of less than one OT site per guide RNA" when using HiFi Cas9 and 20-nt gRNAs [4]. Notably, empirical methods did not identify off-target sites that were not also identified by bioinformatic methods, supporting the potential for "refined bioinformatic algorithms that maintain both high sensitivity and PPV" [4].
Technical Approaches and Innovation
The CRISOT framework represents a significant advancement in addressing bias and overfitting through incorporation of molecular dynamics simulations [27]. This approach derives RNA-DNA molecular interaction fingerprints (CRISOT-FP) from molecular dynamics trajectories, including features such as hydrogen bonding, binding free energies, atom positions, and base pair geometric features [27]. By capturing the underlying biophysical mechanisms of RNA-DNA interaction, CRISOT demonstrates improved generalizability across different CRISPR systems, including base editors and prime editors [27].
Table 2: Technical Approaches to Mitigate Bias and Overfitting in CRISPR AI/ML Models
| Technical Approach | Representative Tools | Methodology | Advantages |
|---|---|---|---|
| Molecular Interaction Fingerprints | CRISOT [27] | Uses MD simulations to derive RNA-DNA interaction features | Captures biophysical mechanisms; more generalizable across systems |
| Hybrid Neural Networks | CRISPR-MCA [39] | Combines multi-scale CNN with multi-head self-attention | Extracts salient information across multiple scales |
| Class Rebalancing | ESB Strategy [39] | Efficiency and Specificity-Based rebalancing for mismatches-only datasets | Addresses extreme class imbalance without introducing artifacts |
| Multi-Feature Integration | CRISTA [39] | Combines genomic content, thermodynamics, and sgRNA-target similarity | Reduces reliance on single feature types that may be biased |
| Transfer Learning | DeepCRISPR [27] | Pre-trains on large datasets before fine-tuning | Improves performance when labeled data is limited |
In head-to-head comparisons using leave-group-out (LGO) and leave-sequence-out (LSO) validation tests, CRISOT-FP demonstrated superior performance compared to state-of-the-art feature encoding methods like Crista_feat, One-hot, and Two-hot encoding, particularly in the more challenging LSO tests where training and testing datasets contained completely different sgRNAs [27].
Experimental Protocols and Methodologies
Benchmarking Experimental Design
The experimental protocol used in comparative studies typically involves several standardized steps to ensure fair evaluation of prediction methods [4]:
gRNA Selection: Researchers select a panel of guide RNAs (typically 10-20) with diverse properties, including different target genes, predicted on-target efficiencies, and varying levels of expected off-target activity. For example, the Cromer et al. (2023) study used 11 gRNAs targeting genes including AAVS1, EMX1, FANCF, HBB, and others, chosen based on disease relevance and inclusion in prior studies [4].
Cell Culture and Editing: Primary cells (such as CD34+ hematopoietic stem and progenitor cells) or cell lines are edited using CRISPR-Cas9 ribonucleoprotein (RNP) complexes, often comparing wild-type Cas9 with high-fidelity variants like HiFi Cas9 to assess specificity differences [4].
Off-target Detection: Multiple empirical methods (e.g., GUIDE-seq, CIRCLE-seq, DISCOVER-Seq) are applied in parallel to identify actual off-target sites experimentally. Next-generation sequencing libraries are prepared for nominated off-target sites.
Computational Prediction: In silico tools are run using the same gRNA sequences, and their predictions are compiled without prior knowledge of empirical results.
Validation: Targeted deep sequencing is performed across all nominated sites (both empirical and computational predictions) to validate editing activity, establishing ground truth data.
Performance Calculation: Sensitivity (ability to identify true off-targets) and positive predictive value (proportion of correct predictions among all predictions) are calculated for each method.
Addressing Class Imbalance: The ESB Strategy
The Efficiency and Specificity-Based (ESB) class rebalancing strategy, introduced specifically for CRISPR off-target prediction, addresses extreme dataset imbalances through a biologically-informed approach [39]. Traditional methods like random undersampling or oversampling can introduce artifacts or remove valuable information [39]. The ESB strategy instead analyzes the location, type, and tolerance of base mismatches within gRNA-target DNA sequences, creating a rebalancing approach based on target efficiency and specificity screening [39].
The protocol involves:
Feature Analysis: Comprehensive analysis of mismatch patterns in off-target datasets, focusing on positional tolerance and type of mismatches.
Efficiency Scoring: Calculation of editing efficiency metrics for different mismatch patterns based on experimental data.
Specificity Screening: Evaluation of which mismatch combinations are most likely to represent true biological off-target events versus artifacts.
Weighted Sampling: Application of sampling weights that prioritize underrepresented but biologically plausible off-target classes based on the efficiency and specificity analysis.
Experimental results demonstrate that the ESB strategy "surpasses five conventional methods in addressing extreme dataset imbalances, demonstrating superior efficacy and broader applicability across diverse models" [39].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Essential Research Reagents for Off-Target Validation Studies
| Reagent/Solution | Function | Application Context |
|---|---|---|
| High-Fidelity Cas9 | Engineered Cas9 variant with reduced off-target activity while maintaining on-target efficiency [4] | All validation studies; provides baseline for optimal specificity |
| CD34+ Hematopoietic Stem/Progenitor Cells | Primary human cells representing clinically relevant model for ex vivo gene therapy [4] | Physiologically relevant editing context with functional DNA repair mechanisms |
| GUIDE-seq Oligos | Double-stranded oligodeoxynucleotides that tag double-strand breaks for genome-wide unbiased identification [4] | Empirical off-target detection in cellular contexts |
| CIRCLE-seq Library Prep Kit | Reagents for circularization for in vitro reporting of cleavage effects by sequencing [4] | Cell-free empirical off-target detection with high sensitivity |
| Site-seq Reagents | Selective enrichment and identification of tagged genomic DNA ends by sequencing [4] | In vitro off-target detection with modified genomic DNA |
| Next-Generation Sequencing Library Prep Kits | Preparation of targeted sequencing libraries for nominated off-target sites [4] | Validation of predicted and empirically detected off-target sites |
| CRISOT-FP Software Suite | Computational framework for generating RNA-DNA interaction fingerprints from molecular dynamics [27] | Advanced in silico prediction with biophysical basis |
| ESB Class Rebalancing Code | Implementation of Efficiency and Specificity-Based rebalancing for machine learning models [39] | Addressing class imbalance in training off-target prediction models |
Integrated Strategies for Mitigating Bias and Overfitting
Technical Mitigation Approaches
The most effective strategies for addressing data bias and overfitting in CRISPR AI/ML models involve a combination of technical approaches tailored to the specific challenges of genomic data:
Pre-processing methods focus on addressing bias problems in training data before model training begins. For CRISPR applications, this includes techniques like the ESB rebalancing strategy [39], synthetic data generation through biologically-informed sequence variation [39], and feature selection that prioritizes molecularly-relevant predictors [27]. These approaches recognize that biased training data creates biased AI systems regardless of algorithm sophistication [40].
In-processing techniques modify the learning algorithms themselves to build fairness directly into models during training. For CRISPR models, this includes adversarial debiasing (where competing networks ensure predictions are independent of confounding factors) [40], regularization methods specifically designed for genomic sequences [39], and architectural choices like the CRISPR-MCA hybrid model that "capitalizes on multi-feature extraction to enhance predictive accuracy" [39].
Post-processing methods adjust AI outputs after the model makes initial decisions to ensure fair results across different sequence types and genomic contexts. These include applying different decision thresholds for different classes of potential off-target sites and calibration techniques that align prediction confidence with empirical observation frequencies [40].
Governance and Validation Frameworks
Beyond technical solutions, comprehensive governance frameworks provide essential oversight for ensuring model fairness and robustness [40]. Effective frameworks include:
Diverse Development Teams: Research consistently shows that homogeneous teams overlook bias issues that diverse groups readily identify [40]. Including team members with different biological expertise (e.g., molecular biologists, computational scientists, clinical researchers) helps identify potential blind spots in model design and interpretation.
Continuous Monitoring: AI systems can develop bias problems after deployment, even when they performed fairly during initial testing [40]. Automated monitoring systems that track performance across different genomic contexts and alert teams to emerging disparities are essential for maintained reliability.
Multi-level Validation: Implementing validation at multiple biological levels—from in silico benchmarks to in vitro confirmation and ultimately in vivo relevance—creates a robust defense against overfitting to specific experimental conditions [4] [5].
The comparative analysis of empirical and in silico off-target prediction methods reveals a evolving landscape where computational approaches are increasingly closing the gap with experimental gold standards. The integration of molecular dynamics simulations, as demonstrated by CRISOT [27], and sophisticated class rebalancing strategies, such as ESB [39], represents a promising direction for addressing fundamental challenges of data bias and overfitting. For researchers and drug development professionals, the optimal path forward leverages the complementary strengths of both approaches: using high-quality empirical data from methods like GUIDE-seq and DISCOVER-Seq to ground truth predictions, while employing advanced in silico tools for comprehensive screening and design optimization. As CRISPR technology advances toward broader therapeutic application, the continued refinement of these AI/ML approaches will be essential for ensuring both safety and efficacy in human genome editing.
Structural characterization of protein–protein interactions (PPIs) across a broad spectrum of scales is fundamental to our understanding of life at the molecular level and for rational drug discovery. The resolution of a protein structure significantly impacts its utility in predicting molecular interactions, understanding biological mechanisms, and identifying off-target effects of therapeutic compounds. In the context of empirical versus in silico off-target prediction methods, the quality of structural data serves as a critical determinant of predictive accuracy. Low-resolution structural modeling provides a necessary approach for modeling large interaction networks, given the significant uncertainties inherent in large biomolecular systems and the high-throughput requirements of the task [43].
The fundamental challenge in structural biology lies in balancing resolution with practical constraints. As noted in foundational literature, "There is nothing worse than a sharp image of a fuzzy concept" [43]. This principle underscores that when high-resolution details are unreliable, lower-resolution representations often provide more biologically meaningful insights. Low-resolution approaches capture essential functional elements without being obscured by potentially inaccurate atomic-level details, making them particularly valuable for modeling complex biological systems where perfect structural data remains unavailable [43].
Comparative Analysis of Structural Determination Methods
Experimental Methods for Structure Determination
Table 1: Comparison of Experimental Protein Structure Determination Methods
| Method | Typical Resolution Range | Throughput | Sample Requirements | Key Applications | Limitations |
|---|---|---|---|---|---|
| X-ray Crystallography | 1.0 - 3.0 Å | Low-Medium | High-purity, crystallizable protein | Detailed atomic structures; ligand binding sites | Requires crystallization; cannot capture dynamics |
| Cryo-EM (Traditional) | 2.5 - 4.5 Å for >50 kDa | Medium | Moderate purity; small amounts | Large complexes; membrane proteins | Challenging for proteins <50 kDa |
| Cryo-EM with Scaffolds | 3.0 - 4.0 Å for small proteins | Low | Engineering of fusion constructs | Small protein targets (e.g., kRasG12C, 19 kDa) | Requires molecular engineering; potential perturbation of native structure |
| NMR Spectroscopy | 1.0 - 3.0 Å (local) | Low | High solubility; isotopic labeling | Solution dynamics; disordered regions | Limited to smaller proteins (<50 kDa) |
Recent advances in cryo-EM have begun to address the long-standing challenge of resolving small proteins. Traditional cryo-EM has been limited to proteins larger than 50 kDa, but innovative scaffolding approaches now enable structural determination of smaller therapeutic targets. For instance, researchers successfully determined the structure of the small protein target kRasG12C (19 kDa) by fusing it to a coiled-coil motif (APH2) recognized by nanobodies, achieving a resolution of 3.7 Å sufficient to visualize the inhibitor drug MRTX849 and GDP in the density map [44]. This approach demonstrates how strategic methodological adaptations can extend the resolution limits of empirical structural biology techniques.
Computational Methods for Structure Prediction
Table 2: Comparison of Computational Protein Structure Prediction Methods
| Method | Typical Resolution (scRMSD) | Throughput | Accuracy Limitations | Key Applications | Notable Tools |
|---|---|---|---|---|---|
| AI-Based Prediction (AlphaFold2) | 1-5 Å (varies by target) | Very High | Static conformations; environmental dependencies | Genome-wide structural coverage; homology gaps | AlphaFold2, ESMFold |
| Sparse Denoising Models | 1-5 Å (designability metrics) | High | Performance degrades >400 residues without optimization | Large protein design; motif scaffolding | SALAD |
| Coarse-Grained Simulations | 5-10 Å (global fold) | Medium | Atomic detail loss; force field approximations | Folding pathways; misfolding mechanisms | Various MD packages |
| Template-Based Docking | 3-8 Å (interface quality) | Medium-High | Template availability; alignment quality | Protein interactome modeling | Comparative modeling |
Computational methods have made remarkable strides, with AI-based systems like AlphaFold2 representing a breakthrough recognized by the 2024 Nobel Prize in Chemistry [45]. However, beneath this apparent success lies a fundamental challenge: these machine learning methods primarily predict static structures from databases of experimentally determined proteins, potentially missing environment-dependent conformational changes crucial for function [45]. The performance of these models is typically evaluated using metrics like self-consistent RMSD (scRMSD) between designed and predicted structures, with scRMSD < 2 Å and pLDDT > 70-80 considered indicators of high confidence [46].
Recent innovations address specific limitations of existing approaches. The SALAD (sparse all-atom denoising) family of models exemplifies progress in generating protein structures with sub-quadratic complexity, enabling efficient generation of diverse and designable backbones for proteins up to 1,000 residues long [46]. By combining sparse attention architectures with denoising diffusion objectives, these models match or outperform state-of-the-art diffusion models while drastically reducing runtime and parameter count [46].
Experimental Protocols for Structural Validation
Cryo-EM with Scaffold Fusion for Small Proteins
Detailed Protocol for kRasG12C Structural Determination [44]:
Construct Design: Fuse kRasG12C to the coiled-coil motif APH2 using a continuous alpha-helical fusion design after deleting the hypervariable C-terminal region including the prenylation site.
Complex Formation: Incubate the kRasG12C-APH2 fusion protein with selected nanobodies (Nb26, Nb28, Nb30, or Nb49) that bind APH2 with high affinity.
Grid Preparation: Apply 3.5 μL of protein complex (0.5 mg/mL concentration) to freshly glow-discharged gold grids (Quantifoil R1.2/1.3, 300 mesh).
Vitrification: Flash-freeze grids in liquid ethane using a Vitrobot Mark IV (4°C, 100% humidity, blot force 10, 4-second blot time).
Data Collection: Acquire images using a 300 keV cryo-electron microscope (Titan Krios) with a K3 direct electron detector at 81,000x magnification, corresponding to a pixel size of 1.07 Å. Collect 5,000 movies with a total electron dose of 50 e-/Ų.
Image Processing: Motion correct and dose-weight frames using MotionCor2. Generate initial models with cryoSPARC, followed by multiple rounds of 2D classification, heterogeneous refinement, and non-uniform refinement.
Model Building: Initially fit the known kRas structure (PDB: 6VJJ) into the density map, followed by iterative manual building in Coot and refinement in Phenix.
This protocol successfully achieved a 3.7 Å resolution structure, enabling clear visualization of the inhibitor MRTX849 and GDP in the electron density map [44].
Validation Metrics for Computational Structures
Designability Assessment Protocol [46]:
Backbone Generation: Generate protein backbone structures using the generative model (e.g., diffusion model, hallucination approach).
Sequence Design: Apply sequence design models (ProteinMPNN, ChromaDesign, or Frame2Seq) to generate amino acid sequences for the designed backbones.
Structure Prediction: Use protein structure predictors (AlphaFold2 or ESMFold) to predict the folded structure of the designed sequences.
Quality Metrics Calculation:
- Compute pLDDT (predicted local distance difference test) to assess prediction confidence.
- Calculate pAE (predicted aligned error) to evaluate positional uncertainty.
- Determine scRMSD (self-consistent RMSD) between the initial design and the predicted structure.
Success Criteria Application: Define successful designs as those with scRMSD < 2 Å and pLDDT > 70 for ESMFold or pLDDT > 80 for AlphaFold2, thresholds shown to produce experimentally viable proteins [46].
Implications for Off-Target Prediction in Drug Discovery
Structural Uncertainty in Empirical vs. In Silico Methods
The resolution of protein structures directly impacts the reliability of off-target prediction in both empirical and computational approaches. Empirical methods for off-target identification—such as GUIDE-Seq, CIRCLE-Seq, and DISCOVER-Seq—operate primarily at the sequence level rather than directly utilizing structural information [4]. However, structural understanding becomes crucial for interpreting the biological consequences of identified off-target effects and designing optimized guide RNAs or small molecules with improved specificity.
In small-molecule drug discovery, in silico target prediction increasingly relies on chemogenomic models that integrate multi-scale information from chemical structures and protein sequences [47]. These methods demonstrate that incorporating protein sequence information significantly improves prediction performance, achieving up to 57.96% of known targets enriched in the top-10 prediction list, representing approximately a 50-fold enrichment over random expectation [47]. However, the absence of high-resolution structural information limits the atomic-level insights necessary for understanding binding mechanics and designing specificity enhancements.
Pathway Analysis: From Structural Determination to Off-Target Prediction
The following workflow diagram illustrates how different resolution structural data feeds into off-target prediction methodologies:
Structural Data in Off-Target Prediction Workflow
This pathway illustrates how both high and low-resolution structural data contribute to complementary approaches for identifying and mitigating off-target effects. While empirical methods primarily rely on sequence information, in silico approaches can leverage structural data at multiple resolution levels to predict potential interactions.
Table 3: Key Research Reagent Solutions for Structural Biology and Off-Target Assessment
| Reagent/Resource | Category | Function | Example Applications |
|---|---|---|---|
| Coiled-coil APH2 module | Protein Scaffold | Enables cryo-EM of small proteins by increasing effective size | Structural studies of small GTPases like kRas (19 kDa) [44] |
| High-affinity Nanobodies | Binding Partners | Stabilize specific protein conformations for structural studies | Cryo-EM structure determination with scaffold fusion [44] |
| DARPin-based Cages | Engineered Scaffold | Provide symmetric environment to stabilize flexible proteins | High-resolution cryo-EM of dynamic proteins [44] |
| SALAD Models | Computational Tool | Sparse denoising for efficient protein structure generation | Designing large proteins up to 1,000 residues [46] |
| AlphaFold2/ESMFold | AI Prediction | Predict protein structures from amino acid sequences | Rapid assessment of protein fold and function [46] |
| Chemogenomic Models | Computational Tool | Integrate chemical and protein data for target prediction | Identifying potential off-target interactions [47] |
| CryoSPARC | Software | Processing pipeline for cryo-EM data | Single-particle analysis and 3D reconstruction [44] |
| ProteinMPNN | Computational Tool | Protein sequence design for given backbones | Generating sequences for designed structures [46] |
Navigating structural uncertainty requires a pragmatic approach that acknowledges the complementary strengths and limitations of both high and low-resolution methods. Low-resolution structural modeling provides an essential tool for modeling large interactomes and addressing biological questions where atomic-level precision is neither necessary nor computationally feasible [43]. The critical insight is that "low resolution does not negate high-resolution" but rather serves as a prerequisite for obtaining high-resolution accuracy through refinement of approximate models [43].
For off-target prediction, the integration of structural information at multiple resolution levels with sequence-based empirical methods offers the most promising path forward. Computational target prediction methods have demonstrated impressive performance, with some models identifying over 57% of known targets in their top-10 predictions [47], but these approaches benefit significantly from structural validation. As structural determination methods continue to advance—particularly for challenging targets like small proteins and flexible complexes—the reliability of both empirical and in silico off-target prediction will correspondingly improve, enabling more effective therapeutic optimization with reduced risk of adverse effects.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) system has revolutionized genome engineering, offering unprecedented opportunities for precise genetic manipulation in both research and therapeutic contexts [22]. This RNA-guided gene-editing technology operates through a complex of Cas nuclease and a single guide RNA (sgRNA) that directs DNA cleavage at specific genomic locations [48]. However, off-target effects—unintended edits at sites with sequence similarity to the target site—remain a significant challenge that can lead to misinterpreted experimental results and serious safety concerns for clinical applications [22] [49].
The persistence of off-target activity stems from the molecular mechanics of CRISPR systems. Cas nucleases can tolerate several mismatches between the sgRNA and genomic DNA, particularly when these mismatches occur in specific positions or patterns [22]. Studies have found that few mismatch DNA sites are potentially recognizable by the sgRNA during the guiding process, with cleavage possible at sites with up to 6 base-pair mismatches [48]. Additional factors including nucleosome occupancy, chromatin accessibility, and binding energy parameters further influence off-target potential [48].
This guide explores the complementary roles of empirical detection methods and in silico prediction tools in characterizing and mitigating off-target effects, with particular focus on how strategic engineering of both gRNA and nuclease components can minimize risks from the initial design phase.
Empirical vs. In Silico Approaches: A Comparative Framework
The scientific community has developed two primary approaches for identifying and quantifying CRISPR off-target activity: experimental detection methods and computational prediction tools. Each approach offers distinct advantages and limitations, with the most comprehensive risk assessment emerging from their integration.
Experimental Detection Methods
Empirical methods directly capture off-target events through biochemical or cell-based assays, providing tangible evidence of nuclease activity across the genome. These techniques vary in their sensitivity, scalability, and biological relevance.
Table 1: Comparison of Major Experimental Off-Target Detection Methods
| Method | Principle | Advantages | Limitations |
|---|---|---|---|
| GUIDE-seq [22] | Integrates double-stranded oligodeoxynucleotides (dsODNs) into double-strand breaks (DSBs) | High sensitivity; cost-effective; low false positive rate | Limited by transfection efficiency |
| CIRCLE-seq [22] | Circularizes sheared genomic DNA followed by in vitro Cas9/sgRNA incubation and sequencing | Ultra-sensitive; minimal background; works without reference genome | In vitro system may not reflect cellular context |
| CHANGE-seq [48] | Scalable, automatable tagmentation-based method for measuring genome-wide Cas9 activity in vitro | High-throughput; applicable to multiple sgRNAs | Limited detection due to experimental apparatus sensitivity |
| Digenome-seq [22] | Digests purified genomic DNA with Cas9/gRNA ribonucleoprotein (RNP) followed by whole-genome sequencing | Highly sensitive; does not require living cells | Expensive; requires high sequencing coverage |
| SITE-seq [22] | Biochemical method with selective biotinylation and enrichment of fragments after Cas9 digestion | Minimal read depth; eliminates background | Lower sensitivity and validation rate |
| DISCOVER-seq [22] | Utilizes DNA repair protein MRE11 for chromatin immunoprecipitation sequencing (ChIP-seq) | Highly sensitive; high precision in cellular contexts | Potential for false positives |
Computational Prediction Tools
In silico methods leverage algorithms to nominate potential off-target sites based on sequence similarity to the intended target. These tools have evolved from simple alignment-based approaches to sophisticated machine learning models incorporating multiple predictive features.
Table 2: Comparison of Computational Off-Target Prediction Tools
| Tool | Algorithm Type | Key Features | Strengths |
|---|---|---|---|
| Cas-OFFinder [22] | Alignment-based | Adjustable sgRNA length, PAM type, mismatch/bulge number | Widely applicable; high tolerance for variations |
| FlashFry [22] | Alignment-based | High-throughput; provides GC content and on/off-target scores | Fast analysis of hundreds of thousands of targets |
| CFD [22] | Scoring-based | Based on experimentally validated dataset | Position-specific mismatch weighting |
| CCTop [22] | Scoring-based | Considers distances of mismatches to PAM | User-friendly web interface |
| DeepCRISPR [22] | Deep learning | Incorporates both sequence and epigenetic features | Enhanced prediction accuracy through neural networks |
| crispAI [48] | Neural network | Provides uncertainty estimates using Zero Inflated Negative Binomial model | Quantifies prediction confidence; superior performance |
Integrated Workflow for Comprehensive Off-Target Assessment
The most robust approach to off-target assessment combines both empirical and computational methods in a complementary workflow. Empirical data validates and refines computational predictions, while in silico tools help prioritize sites for experimental validation.
gRNA Engineering Strategies for Enhanced Specificity
Strategic design of guide RNA represents the first and most accessible approach for minimizing off-target effects. Multiple parameters can be optimized during gRNA design to enhance specificity while maintaining on-target activity.
gRNA Length Modification
Truncated gRNAs with shorter complementarity regions demonstrate reduced off-target activity while preserving on-target efficiency. Standard 20-nucleotide guides can be shortened to 17-18 nucleotides, decreasing non-specific binding energy while maintaining sufficient specificity for target recognition.
Experimental Protocol: Evaluating Truncated gRNA Efficacy
- Design: Create a series of gRNAs with progressively shorter complementarity regions (20nt, 18nt, 17nt) targeting the same genomic locus
- Synthesis: Chemically synthesize or in vitro transcribe truncated gRNAs
- Delivery: Transfect gRNAs with Cas9 nuclease into target cells using appropriate method (lipofection, electroporation)
- Assessment: Measure on-target efficiency via T7E1 assay or next-generation sequencing (NGS)
- Off-target Evaluation: Perform GUIDE-seq or targeted sequencing of predicted off-target sites
- Validation: Compare editing profiles of truncated versus full-length gRNAs
Chemical Modifications and Enhanced Specificity Designs
Chemical modifications to gRNA backbone and termini can improve nuclease resistance and enhance specificity. Additionally, specialized gRNA architectures such as double-guide RNAs and extended sgRNAs (esgRNAs) offer alternative approaches to reduce off-target effects.
Nuclease Engineering for Reduced Off-Target Activity
Protein engineering of Cas nucleases has yielded variants with dramatically improved specificity profiles. These engineered nucleases maintain robust on-target activity while exhibiting reduced tolerance for mismatched target sequences.
High-Fidelity Cas Variants
Multiple research groups have developed enhanced specificity mutants through rational design and directed evolution approaches. These variants typically incorporate mutations that destabilize Cas binding to mismatched targets.
Table 3: Engineered High-Fidelity Cas Nuclease Variants
| Nuclease | Parent | Key Mutations | Specificity Improvement | PAM Sequence |
|---|---|---|---|---|
| SpCas9-HF1 [22] | SpCas9 | K848A, K1003A, R1060A | Reduced off-targets while maintaining on-target | NGG |
| eSpCas9(1.1) [22] | SpCas9 | K848A, K1003A, R1060A | Enhanced specificity through altered binding kinetics | NGG |
| SpCas9-NG [22] | SpCas9 | R1335V, L1111R, etc. | Relaxed PAM requirement (NG) with maintained specificity | NG |
| hfCas12Max [50] | Cas12i | Engineered variant | High-fidelity with simplified PAM requirement | TN and/or TNN |
| xCas9 [22] | SpCas9 | Multiple mutations | Broad PAM recognition with improved specificity | NG, GAA, GAT |
PAM Specificity and Novel Nuclease Discovery
The Protospacer Adjacent Motif (PAM) requirement represents a fundamental constraint on CRISPR targeting, but also provides an opportunity for specificity enhancement. Natural and engineered Cas variants with altered PAM requirements can expand targetable genomic space while reducing off-target potential.
Experimental Protocol: Characterizing Novel Nuclease Specificity
- Library Design: Create a diverse sgRNA library targeting known genomic sites with varying mismatch patterns
- Screening: Express novel nuclease variant in target cells and deliver sgRNA library
- Deep Sequencing: Perform targeted amplicon sequencing of both on-target and predicted off-target sites
- Activity Profiling: Quantify editing efficiency at each site using computational analysis tools
- Specificity Scoring: Calculate specificity scores based on ratio of on-target to off-target activity
- Validation: Confirm top findings using orthogonal methods (GUIDE-seq, CIRCLE-seq)
Table 4: Natural Cas Nucleases and Their PAM Requirements
| Nuclease | Organism Source | PAM Sequence (5' to 3') | Notes |
|---|---|---|---|
| SpCas9 [50] | Streptococcus pyogenes | NGG | Most widely used; standard for comparison |
| SaCas9 [50] | Staphylococcus aureus | NNGRRT or NNGRRN | Compact size advantageous for viral delivery |
| NmeCas9 [50] | Neisseria meningitidis | NNNNGATT | Longer PAM increases specificity |
| Cas12a (Cpf1) [50] | Lachnospiraceae bacterium | TTTV | T-rich PAM; different cleavage pattern |
| Cas12b [50] | Alicyclobacillus acidiphilus | TTN | Thermostable variant available |
Advanced Dual-Targeting Systems and Library Design
Innovative approaches that combine multiple CRISPR modalities or optimize screening library design offer additional strategies for reducing off-target effects while maintaining screening sensitivity.
Dual-Targeting Approaches
Dual-targeting CRISPR systems utilize two distinct sgRNAs to enhance specificity and efficiency. Recent research demonstrates that dual CRISPRko approaches can create deletions between target sites, potentially increasing knockout efficiency, though they may trigger heightened DNA damage response [17]. More advanced systems like CRISPRgenee combine gene knockout with epigenetic repression in a single coordinated system [51].
Mechanism of CRISPRgenee System:
- Simultaneously targets Cas9 nuclease activity and epigenetic repression to the same locus
- Uses truncated sgRNAs (15nt) to maintain epigenetic silencing without DNA cleavage
- Combines KRAB-mediated transcriptional repression with nuclease activity
- Demonstrates improved depletion efficiency and reduced sgRNA performance variance [51]
Optimized sgRNA Library Design
Benchmark studies comparing genome-wide CRISPR libraries reveal that smaller, more focused libraries can perform as well or better than larger conventional libraries when guides are chosen according to principled criteria [17] [52]. The Vienna library, which selects guides based on VBC scores, demonstrates that libraries with only 3 guides per gene can achieve strong depletion of essential genes while reducing off-target potential through careful design [17].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of off-target minimization strategies requires appropriate selection of research reagents and tools. The following table summarizes key solutions for designing and evaluating specific CRISPR experiments.
Table 5: Essential Research Reagents for Off-Target Assessment
| Reagent/Tool | Function | Application Context | Example Products |
|---|---|---|---|
| High-Fidelity Cas Nucleases [22] | Engineered variants with reduced off-target activity | All CRISPR applications requiring high specificity | SpCas9-HF1, eSpCas9(1.1) |
| CHANGE-seq Kit [48] | In vitro off-target detection using tagmentation | Genome-wide off-target profiling | CHANGE-seq Kit |
| GUIDE-seq Oligos [22] | Double-stranded oligodeoxynucleotides for DSB capture | Comprehensive off-target mapping in cells | GUIDE-seq dsODN |
| CRISPR Library Sets [17] | Pre-designed sgRNA collections for specific applications | Functional genomic screens | Vienna Library, Brunello Library |
| crispAI Software [48] | Neural network-based off-target prediction with uncertainty estimates | Computational off-target risk assessment | crispAI GitHub Package |
| Cas-OFFinder Tool [22] | Genome-wide search for potential off-target sites | Initial sgRNA design and risk evaluation | Cas-OFFinder Web Tool |
Minimizing off-target activity in CRISPR applications requires a multifaceted approach that begins with strategic design decisions. The most effective outcomes emerge from the integration of computational prediction with empirical validation, informed by continuous advances in both gRNA and nuclease engineering. As CRISPR technology progresses toward therapeutic applications, robust off-target assessment becomes increasingly critical. By implementing the engineering strategies and assessment methods outlined in this guide, researchers can significantly enhance the specificity of their genome editing experiments while maintaining high on-target efficiency. The evolving landscape of CRISPR engineering—including continued development of novel nucleases with distinct PAM specificities, enhanced prediction algorithms that incorporate epigenetic features, and innovative dual-targeting approaches—promises to further narrow the gap between experimental intention and genomic outcome.
The expansion of biological data has created a critical need for sophisticated data curation practices, particularly in high-stakes fields like drug discovery and therapeutic genome editing. A central theme in modern bioinformatics is the interplay between empirical methods (hypothesis-driven, experimental) and in silico methods (discovery-based, computational) for data generation and validation [53]. While empirical data has traditionally been perceived as more reliable, evaluations find that literature curation can be error-prone and of lower quality than commonly assumed [53]. Conversely, purely computational approaches may miss critical biological context. This comparison guide examines best practices for curating datasets that leverage the strengths of both approaches, with special focus on incorporating negative data and establishing confidence metrics for biological interactions, drawing from recent advances in protein interaction databases, drug-target resources, and CRISPR off-target prediction platforms.
Protein-Protein Interaction Databases
Literature-curated protein-protein interaction (PPI) datasets face significant challenges in completeness and reliability. Surprisingly, more than 75% of yeast PPIs and 85% of human PPIs in curated databases are supported by only a single publication, with only a small fraction (5% or less) described in ≥3 publications [53]. This lack of independent validation raises concerns about data reliability. Different major databases (MINT, IntAct, and DIP) show surprisingly low overlaps of curated PPIs and PubMed coverage, suggesting curation is far from comprehensive [53].
Table 1: Coverage and Multi-Support Analysis of Literature-Curated PPI Datasets
| Organism | Total PPIs | Supported by Single Publication | Supported by ≥3 Publications | Supported by ≥5 Publications |
|---|---|---|---|---|
| Yeast | 11,858 | 75% | 5% | 2% |
| Human | 4,067 | 85% | 5% | 1% |
| Arabidopsis | Not specified | 93% | 1% | 0.1% |
Drug-Target Interaction Databases
The HCDT 2.0 database represents a significant advancement in drug-target interaction curation, containing 1,284,353 curated interactions across multiple types: 1,224,774 drug-gene pairs, 11,770 drug-RNA mappings, and 47,809 drug-pathway links [54]. A crucial innovation in HCDT 2.0 is the systematic integration of 38,653 negative drug-target interactions across 26,989 drugs and 1,575 genes, defined by experimental binding affinity measurements (Ki/Kd/IC50/EC50/AC50/Potency >100 μM) [54]. This addresses a critical gap in most interaction databases that primarily capture positive interactions.
Table 2: HCDT 2.0 Database Composition and Interaction Types
| Interaction Type | Number of Interactions | Entity Coverage | Key Filtering Criteria |
|---|---|---|---|
| Drug-Gene | 1,224,774 | 678,564 drugs × 5,692 genes | Ki, Kd, IC50, EC50 ≤10 μM |
| Drug-RNA | 11,770 | 316 drugs × 6,430 RNAs | Experimentally validated, human origin |
| Drug-Pathway | 47,809 | 6,290 drugs × 3,143 pathways | Experimentally validated |
| Negative DTIs | 38,653 | 26,989 drugs × 1,575 genes | Binding affinity >100 μM |
CRISPR Off-Target Prediction Tools
Comparative studies of CRISPR off-target discovery methods reveal important insights for data curation. When comparing in silico tools (COSMID, CCTop, Cas-OFFinder) and empirical methods (CHANGE-Seq, CIRCLE-Seq, DISCOVER-Seq, GUIDE-Seq, SITE-Seq) after editing hematopoietic stem and progenitor cells, researchers found that empirical methods did not identify off-target sites that were not also identified by bioinformatic methods [4]. COSMID, DISCOVER-Seq, and GUIDE-Seq attained the highest positive predictive value (PPV), suggesting that refined bioinformatic algorithms could maintain both high sensitivity and PPV [4].
Experimental Protocols and Methodologies
High-Confidence Interaction Curation Protocol
The HCDT 2.0 database employs a stringent methodology for data collection, curation, and integration to ensure precision and reliability [54]:
Multi-Source Data Aggregation: Collect data from 9 specialized databases for drug-gene interactions, 6 databases for drug-RNA interactions, and 5 databases for drug-pathway interactions.
Strict Filtering Criteria:
- For gene data: Ki, Kd, IC50, and EC50 with at least one ≤10 micromoles
- For RNA data: Experimental validation with human origin requirement
- For pathway data: Experimentally validated relationships rather than predicted
Standardized Identifier Mapping:
- Drugs: SMILES, IUPAC name, INCH identifiers
- Genes: Gene symbol, Entrez ID, Ensemble ID, or UniProt ID mapped to HGNC
- RNAs: Ensemble ID as primary identifier
- Pathways: REACTOMEID, KEGGHSAID, SMPDB_ID
Comprehensive Classification:
- Genes: Protein-coding, non-coding RNA, pseudogenes, unknown function
- RNAs: miRNA, lncRNA, general RNA, circRNA, piRNA
- Pathways: Categorized by source database
CRISPR Off-Target Validation Workflow
A comprehensive study comparing off-target prediction methods utilized this rigorous experimental protocol [4]:
Cell System: Primary human CD34+-purified hematopoietic stem and progenitor cells (HSPCs) edited ex vivo using clinically relevant RNP delivery.
Editing Conditions: 11 different gRNAs complexed with Cas9 protein (both high-fidelity and wild-type versions) with 20-nt and 18-nt spacer lengths.
Off-Target Nomination: Multiple in silico tools (COSMID, CCTop, Cas-OFFinder) and empirical methods (CHANGE-Seq, CIRCLE-Seq, DISCOVER-Seq, GUIDE-Seq, SITE-Seq) were applied in parallel.
Validation: Targeted next-generation sequencing of all nominated off-target sites to classify as true or false positives.
Performance Metrics: Calculation of sensitivity and positive predictive value for each method.
Incorporating Population Genetic Variability in Off-Target Prediction
Advanced off-target prediction must account for genetic variability across populations [55]:
Variant Integration: Analysis of polymorphic sites within potential off-target sequences using 1000 Genomes phase 3 data (2,504 individuals).
PAM Disruption Analysis: Evaluation of how polymorphic sites may create or disrupt PAM sequences (NGG).
Population-Specific Scoring: Calculation of cleavage probabilities using CFD score while considering population allele frequencies.
Functional Context Assessment: Annotation of off-target sequences as genic, intergenic, or pseudogene regions.
Data Curation Workflow: High-confidence interaction curation involves multiple validation stages before FAIR publication.
Visualization of Methodologies and Relationships
Empirical vs. In Silico Method Characteristics
Method Comparison: Empirical and in silico approaches exhibit complementary strengths and limitations [53].
CRISPR Off-Target Validation Workflow
Off-target Validation: Combined empirical and computational methods improve prediction accuracy [4].
Table 3: Key Research Reagent Solutions for Data Curation and Validation Studies
| Resource | Function | Application Context |
|---|---|---|
| High-Fidelity Cas9 | Engineered nuclease with reduced off-target activity | CRISPR therapeutic safety assessment [4] |
| GUIDE-Seq | Unbiased in vitro off-target detection | Genome-wide identification of CRISPR off-target sites [4] |
| CIRCLE-Seq | In vitro circularization for off-target detection | Sensitive identification of potential off-target sites [4] |
| HCDT 2.0 Database | Comprehensive drug-target interaction resource | Drug discovery and repurposing, adverse event prediction [54] |
| COSMID | CRISPR Off-target Sites with Mismatches, Insertions, and Deletions | Specific CRISPR off-target prediction with stringent criteria [55] |
| CRISOT Tool Suite | RNA-DNA interaction fingerprint for off-target prediction | Genome-wide CRISPR off-target prediction and sgRNA optimization [27] |
| BioGRID | Protein-protein interaction repository | Literature-curated PPI data for network analysis [53] |
The comparative analysis reveals that neither purely empirical nor exclusively in silico methods suffice for comprehensive data curation. Rather, the most robust practices integrate both approaches while emphasizing negative data incorporation and multi-support validation. Key findings indicate that:
Database comprehensiveness remains challenging, with major protein interaction databases showing surprisingly low overlap despite years of curation [53].
Negative data integration, as demonstrated in HCDT 2.0, addresses critical gaps in interaction databases and improves predictive modeling [54].
Combined computational and empirical validation, as seen in CRISPR off-target studies, provides higher confidence than either approach alone [4] [27].
Population genetic variability must be considered in curation practices, as polymorphisms significantly impact interaction predictions and editing outcomes [55].
The progression toward FAIR (Findable, Accessible, Interoperable, Reusable) data principles, coupled with advanced machine learning approaches that leverage both positive and negative examples, represents the most promising path forward for biological data curation [56]. These practices will be essential for accelerating drug discovery and ensuring the safety of emerging therapeutic modalities like CRISPR-based gene editing.
The integration of in silico technologies with traditional experimental methods represents a paradigm shift in biomedical research, particularly in drug discovery and development. This hybrid approach leverages computational power to predict biological outcomes while relying on experimental data for validation, creating a synergistic cycle that enhances both efficiency and reliability. The core premise of these hybrid workflows is to address the critical challenge of process-model mismatch (PMM), where discrepancies emerge between computational predictions and actual biological processes [57]. By continuously cross-validating computational findings with early-stage experimental results, researchers can refine models, improve predictive accuracy, and accelerate the translation of discoveries from bench to bedside.
The evolution from primarily in vivo (within living organisms) and in vitro (in controlled laboratory environments) methods to advanced in silico (computer-simulated) approaches has revolutionized research methodologies [58]. This transition is particularly relevant in the context of off-target prediction for therapeutic development, where the stakes for accuracy are extraordinarily high. Whether developing small-molecule drugs or CRISPR-based gene therapies, researchers must navigate the delicate balance between efficacy and safety, making the precise identification of off-target effects a critical determinant of success [59] [5].
Comparative Analysis of Hybrid Workflow Performance
The following table summarizes quantitative performance data for hybrid in silico/experimental workflows across various applications, demonstrating their tangible benefits in preclinical research and development.
Table 1: Performance Metrics of Hybrid In Silico/Experimental Workflows
| Application Area | Reported Metric | Performance Outcome | Reference/Model |
|---|---|---|---|
| Drug Discovery Timeline | Time to Market | Reduction of several years compared to traditional methods [58] | InSilicoTrials Case Study |
| Clinical Trial Efficiency | Patient Enrollment | 256 fewer patients required in clinical study [58] | Medtronic Implementation |
| Economic Impact | Cost Savings | $10 million saved due to reduced patient numbers and early market dominance [58] | Medtronic Implementation |
| Cancer Drug Discovery | Binding Energy (against AKT1) | -11.4 kcal/mol for ELRC-LC hybrid, indicating stronger binding than native compounds [60] | Curcumin-Resveratrol Hybrid Study |
| Toxicity Prediction | LD₅₀ Prediction Accuracy | Random Forest model achieved r² = 0.8410, RMSE = 0.1112 [61] | ADME-Tox Profiling Study |
| Bioprocess Optimization | Fatty Acid Production | Improved yield through mitigation of process-model mismatch [57] | HISICC (E. coli FA3 strain) |
Experimental Protocols for Hybrid Workflow Validation
Protocol: Molecular Docking and Dynamics for Hybrid Molecule Validation
This protocol outlines the methodology for computationally designing and experimentally validating hybrid molecules with enhanced therapeutic properties, as demonstrated in the development of curcumin-resveratrol hybrids for cancer therapy [60].
Step 1: Computational Design and Geometry Optimization
- Methodology: Candidate hybrid structures are designed using molecular software (e.g., Avogadro v1.2.0). Geometry optimization is performed using Density Functional Theory (DFT) to achieve stable electronic configurations.
- Key Analyses: Characterize electronic properties through Frontier Molecular Orbital (FMO), Molecular Electrostatic Potential (MEP), and Fourier Transform Infrared (FTIR) analyses [60].
Step 2: Molecular Docking against Target Proteins
- Methodology: Perform molecular docking to calculate binding energies of hybrids against specific oncogenic targets (e.g., AKT1, MAPK, STAT3). Compare results to native compounds (curcumin, resveratrol) and reference inhibitors.
- Output: Binding affinity measurements (in kcal/mol) identifying promising candidates [60].
Step 3: Molecular Dynamics (MD) Simulations
- Methodology: Submit best-fitting complexes from docking to 100 ns Molecular Dynamics (MD) simulations to evaluate conformational stability and binding interactions over time.
- Validation Metric: Calculate binding free energies using the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) method [60].
Step 4: Experimental Correlation
- Methodology: Compounds with favorable in silico profiles proceed to in vitro testing (e.g., resazurin cell viability assay, ROS measurement in cancer cell lines like A549, MCF-7, MDA-MB-231) [60].
- Cross-Validation: Compare predicted binding affinities with experimentally measured IC₅₀ values and apoptotic activity.
Protocol: ADME-Tox Profiling with Machine Learning Integration
This protocol details an integrated computational approach for predicting absorption, distribution, metabolism, excretion, and toxicity (ADME-Tox) profiles early in the drug discovery process, combining in silico tools with machine learning [61].
Step 1: Compound Preparation and Descriptor Calculation
- Methodology: Optimize chemical structures using the MMFF94 force field. Calculate key ADME-Tox descriptors (Log P, Log S, Caco-2 permeability, CYP450 interactions, hERG inhibition, LD₅₀, DILI) using platforms like SwissADME and PreADMET [61].
Step 2: Data Analysis and Pattern Recognition
- Methodology: Perform statistical analyses including Pearson correlation and Principal Component Analysis (PCA) to identify trends and relationships between molecular properties.
- Clustering: Apply hierarchical clustering and construct cosine similarity networks to group structurally related compounds [61].
Step 3: Machine Learning Model Development
- Methodology: Implement Random Forest regression to predict key toxicity endpoints (e.g., LD₅₀). Validate model performance using five-fold cross-validation to ensure robustness and prevent overfitting [61].
Step 4: Experimental Correlation and Model Refinement
Protocol: Hybrid In Silico/In-Cell Control for Bioprocess Optimization
This protocol describes the implementation of a Hybrid In Silico/In-Cell Controller (HISICC) to address process-model mismatches in engineered microbial bioprocessing, exemplified in fatty acid production using E. coli [57].
Step 1: System Modeling and In Silico Controller Design
- Methodology: Develop mathematical models of engineered strains (e.g., FA2, FA3 for fatty acid production) based on known metabolic pathways and genetic circuit behavior.
- Controller Design: Design in silico feedforward controllers to optimize process inputs (e.g., inducer concentration like IPTG) that regulate key enzyme expression (e.g., acetyl-CoA carboxylase) [57].
Step 2: Implementation of Intracellular Biosensing
- Methodology: Engineer microbial strains with genetically encoded feedback controllers (e.g., FA3 strain with malonyl-CoA-responsive transcription factor FapR). These in-cell feedback controllers autonomously adjust enzyme expression in response to metabolite levels [57].
Step 3: Hybrid Control Operation
- Methodology: The in silico controller sets optimal initial induction parameters, while the in-cell controller makes real-time adjustments based on actual intracellular conditions during the bioprocess.
- Outcome Measurement: Monitor key metrics including enzyme concentrations, metabolite levels, cell growth, and final product yields [57].
Step 4: Handling Process-Model Mismatch (PMM)
- Methodology: When PMM occurs (e.g., unexpectedly rapid enzyme accumulation), the HISICC automatically adjusts to slow induction before cytotoxic levels are reached, thereby improving overall yield compared to non-hybrid systems [57].
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 2: Key Research Reagents and Computational Platforms for Hybrid Workflows
| Tool/Reagent | Type | Primary Function | Example Application |
|---|---|---|---|
| Avogadro Software | Computational Chemistry | Molecular design and editing | Designing curcumin-resveratrol hybrid molecules [60] |
| SwissADME/PreADMET | ADME-Tox Prediction | In silico pharmacokinetic and toxicity profiling | Predicting Log P, Log S, CYP450 interactions for compound prioritization [61] |
| Engineered E. coli FA3 Strain | Biological System | Fatty acid production with malonyl-CoA biosensing | Implementing HISICC for bioprocess optimization [57] |
| FapR/FR1 Genetic Circuit | Biosensor Device | Detects malonyl-CoA and regulates gene expression | Autonomous feedback control of ACC expression in FA3 strain [57] |
| PyRx/Discovery Studio | Molecular Docking | Predicting ligand-protein interactions | Identifying potential TLK2 kinase inhibitors for breast cancer [61] |
| Random Forest Algorithm | Machine Learning | Predictive modeling of complex biological endpoints | LD₅₀ toxicity prediction with high accuracy (r² = 0.8410) [61] |
| Patient-Derived Xenografts (PDXs) | Experimental Model | In vivo validation of drug candidates | Cross-validating AI predictions of tumor response [62] |
The integration of in silico predictions with early-stage experimental data represents a fundamental advancement in biomedical research methodology. As demonstrated across multiple applications—from cancer drug discovery to microbial metabolic engineering—hybrid workflows consistently enhance efficiency, reduce costs, and improve predictive accuracy compared to traditional single-approach methods. The critical advantage of these frameworks lies in their capacity for perpetual refinement, where discrepancies between predictions and experimental outcomes become opportunities for model improvement rather than单纯的 failures [58].
The future trajectory of hybrid validation will likely involve increased incorporation of artificial intelligence and multi-scale modeling, integrating data from molecular, cellular, and tissue levels to create more comprehensive biological simulations [62]. Furthermore, as regulatory agencies like the FDA continue to endorse Model-Informed Drug Development (MIDD) approaches, the adoption of these hybrid methodologies is expected to accelerate, potentially transforming how therapies are developed and validated [58]. For researchers navigating the complex landscape of off-target prediction and therapeutic safety, these hybrid workflows offer a robust framework for balancing innovation with responsibility, ultimately accelerating the delivery of safer, more effective treatments to patients.
Benchmarking, Validation, and Regulatory Considerations for Real-World Application
In the rapidly advancing field of computational biology, the development of in silico prediction methods has dramatically outpaced the establishment of standardized validation frameworks. This discrepancy poses significant challenges for researchers, scientists, and drug development professionals who rely on these tools for critical decisions. The core thesis distinguishing empirical validation—relying on physical experimentation and observation—from purely in silico approaches—utilizing computational models and simulations—forms the central context for this guide. As noted by Nature Computational Science, even computational-focused research often requires experimental validation to verify reported results and demonstrate practical usefulness [63]. This guide provides a comprehensive comparison of validation frameworks, synthesizing current methodologies, quantitative performance data, and experimental protocols to establish benchmarks for assessing computational prediction tools in biomedical research.
Comparative Analysis of Validation Frameworks
Core Principles: VVUQ and Beyond
A robust framework for validating computational predictions rests on the triad of Verification, Validation, and Uncertainty Quantification (VVUQ). In precision medicine, these processes are essential for ensuring the safety and efficacy of digital twins and other computational tools [64].
- Verification answers "Did we build the system right?" It involves ensuring that software components perform as expected through code solution verification and software quality engineering practices [64].
- Validation addresses "Did we build the right system?" It tests models for applicability and accuracy against real-world experimental data across various scenarios [64].
- Uncertainty Quantification (UQ) formally tracks uncertainties throughout model calibration, simulation, and prediction, distinguishing between epistemic uncertainties (incomplete knowledge) and aleatoric uncertainties (natural variability) [64].
The emerging concept of dynamic validation presents particular challenges for digital twins, which are continuously updated with new data. This necessitates more flexible and iterative temporal validation approaches compared to traditional static models [64].
Domain-Specific Validation Challenges
Different biological domains present unique validation challenges and requirements:
Spatial Prediction Problems: Weather forecasting and air pollution mapping exemplify spatial prediction tasks where traditional validation methods can fail dramatically. MIT researchers demonstrated that common validation techniques make inappropriate assumptions about spatial data being independent and identically distributed. Their proposed solution incorporates a spatial regularity assumption, where validation data and test data are assumed to vary smoothly across space, resulting in more accurate validations for problems like wind speed prediction and air temperature forecasting [65].
Allosteric Site Prediction: The field of allosteric drug discovery faces distinct validation hurdles due to limited evolutionary conservation of allosteric sites, conformational flexibility, and transient pockets. Computational strategies combining machine learning, molecular dynamics, and network-based approaches require specialized validation against experimental structural biology techniques like X-ray crystallography and cryo-EM, though these methods themselves face challenges in capturing transient states [66].
Protein Structure Prediction: The revolutionary AlphaFold2 system has necessitated new validation approaches. Comprehensive analyses comparing AF2-predicted and experimental nuclear receptor structures reveal that while AF2 achieves high accuracy for stable conformations with proper stereochemistry, it shows limitations in capturing flexible regions, ligand-binding pockets, and functionally important conformational diversity. Validation metrics include root-mean-square deviations, secondary structure elements, domain organization, and ligand-binding pocket geometry [67].
Table 1: Validation Framework Comparison Across Domains
| Domain | Primary Validation Methods | Key Metrics | Unique Challenges |
|---|---|---|---|
| Spatial Predictions [65] | Spatial regularity validation, holdout validation | Prediction accuracy, Spatial smoothness | Inappropriate independence assumptions, Location-based statistical variations |
| Allosteric Site Prediction [66] | Molecular dynamics, Network analysis, Machine learning validation | Cryptic pocket identification, Communication pathways | Transient pockets, Conformational flexibility, Limited conservation |
| Protein Structure Prediction [67] | Experimental structure comparison, pLDDT scoring | RMSD, Secondary structure accuracy, Pocket volumes | Capturing conformational diversity, Flexible regions, Ligand binding sites |
| Variant Effect Prediction [18] | Experimental mutagenesis, Cross-validation, Functional enrichment | Accuracy, Precision, Recall, F1-score | Data scarcity, Generalizability, Regulatory region interpretation |
| Digital Twins in Medicine [64] | VVUQ, Dynamic validation, Clinical comparison | Predictive accuracy, Clinical relevance, Uncertainty bounds | Continuous model updating, Clinical translation, Trust establishment |
Experimental Protocols for Validation
Spatial Prediction Validation Methodology
The MIT validation technique for spatial predictions employs a systematic protocol [65]:
- Data Preparation: Collect spatial data with explicit location information for both validation and test datasets.
- Assumption Testing: Evaluate whether traditional independence assumptions are violated by analyzing spatial correlations in the data.
- Smoothness Validation: Apply the spatial regularity assumption that data varies smoothly across geographic space.
- Predictor Assessment: Input the predictor, target prediction locations, and validation data into the validation algorithm.
- Accuracy Estimation: The method automatically estimates prediction accuracy for specified locations, accounting for spatial relationships.
This protocol was validated through experiments with real and simulated data, including predicting wind speed at Chicago O'Hare Airport and air temperature at five U.S. metro locations [65].
Machine Learning Model Validation
For sequence-based AI models predicting variant effects, the validation protocol involves [18]:
- Data Segmentation: Partition data into training, validation, and test sets, ensuring representative distribution of variants.
- Cross-Validation: Implement k-fold cross-validation to assess model generalizability across different data subsets.
- Performance Metrics: Calculate accuracy, precision, recall, and F1-score against experimental data.
- External Validation: Test model predictions on independent datasets not used in training.
- Functional Analysis: Perform enrichment analyses to validate biological relevance of predictions.
- Experimental Verification: Where possible, confirm predictions through mutagenesis screens or functional assays.
Digital Twin Validation Framework
Validation of digital twins in precision medicine requires a comprehensive approach [64]:
Verification Phase:
- Code verification through software quality engineering practices
- Solution verification assessing mathematical model discretization convergence
- Algorithm correctness validation
Validation Phase:
- Comparison against clinical data across diverse patient populations
- Scenario testing for specific conditions (e.g., cancer types, treatment regimens)
- Predictive accuracy assessment for health trajectories
Uncertainty Quantification:
- Parameter uncertainty analysis through sensitivity testing
- Model form uncertainty evaluation
- Propagation of uncertainty through simulations
- Confidence bound establishment for predictions
Performance Data and Quantitative Comparisons
Validation Method Efficacy
Table 2: Quantitative Performance Comparison of Validation Methods
| Method | Application Context | Reported Performance | Limitations |
|---|---|---|---|
| Traditional Spatial Validation [65] | Weather forecasting, Pollution mapping | Can be "substantively wrong" due to inappropriate assumptions | Fails when data are not independent and identically distributed |
| MIT Spatial Regularity Approach [65] | Wind speed, Temperature forecasting | More accurate than two common classical methods | Requires spatial smoothness assumption |
| Deep Reinforcement Learning (ncRNADS) [68] | ncRNA-disease associations in breast cancer | 96.20% accuracy, 96.48% precision, 96.10% recall, 96.29% F1-score | Specific to ncRNA classification, requires large feature set |
| AlphaFold2 Structural Prediction [67] | Nuclear receptor structure modeling | High stereochemical quality but underestimates ligand-binding pocket volumes by 8.4% on average | Misses functional asymmetry in homodimeric receptors |
| Sequence Model Variant Prediction [18] | Plant breeding variant effect prediction | Generalizes across genomic contexts but accuracy depends heavily on training data | Limited by data scarcity, especially for regulatory sequences |
Domain-Specific Performance Metrics
Allosteric Site Prediction: Machine learning approaches for allosteric site prediction demonstrate varying performance depending on feature selection and model architecture. The integration of molecular dynamics simulations enhanced by advanced sampling algorithms has improved identification of cryptic binding pockets, though high computational costs remain a limitation [66].
Variant Effect Prediction: Unsupervised models in comparative genomics, such as those based on evolutionary conservation, show promise for identifying deleterious variants. However, their accuracy is constrained by limited availability of related genomes and difficulties in generating homologous alignments [18].
Signaling Pathways and Workflows
Integrated Computational Validation Framework
Validation Workflow Integration
Allosteric Prediction Computational Pipeline
Allosteric Prediction Pipeline
Research Reagent Solutions
Table 3: Essential Research Resources for Validation Experiments
| Resource/Platform | Type | Primary Function in Validation | Access Information |
|---|---|---|---|
| Protein Data Bank (PDB) [67] | Database | Provides experimental structures for benchmarking computational predictions | https://www.rcsb.org/ |
| AlphaFold Protein Structure Database [67] | Database | Source of AI-predicted structures for comparison with experimental data | https://alphafold.ebi.ac.uk/ |
| GPCRmd database [66] | MD Repository | Offers molecular dynamics trajectories for validating dynamic predictions | https://gpcrmd.org/ |
| Cancer Genome Atlas [63] | Database | Provides genomic data for validating variant effect predictions | https://www.cancer.gov/ccg/research/genome-sequencing/tcga |
| MorphoBank [63] | Database | Evolutionary biology data for validating phylogenetic predictions | https://morphobank.org/ |
| High Throughput Experimental Materials Database [63] | Database | Materials science data for validating computational material predictions | https://htem.nrel.gov/ |
| PubChem [63] | Database | Chemical compound information for validating molecular design predictions | https://pubchem.ncbi.nlm.nih.gov/ |
The establishment of a gold standard for validating computational predictions requires a multifaceted approach that integrates empirical validation with sophisticated in silico techniques. As computational methods continue to advance, validation frameworks must evolve correspondingly, particularly through dynamic validation approaches for continuously updated models like digital twins [64]. The integration of machine learning, molecular dynamics, and network-based approaches demonstrates the power of combined methodologies for addressing complex biological questions [66]. However, significant challenges remain in data scarcity, model generalizability, computational expenses, and the translation of computational predictions to clinically actionable tools. Moving forward, the field must prioritize the development of standardized validation protocols, sharing of high-quality experimental datasets, and robust uncertainty quantification to build trust in computational predictions across research and clinical applications.
The advancement of CRISPR/Cas9 genome editing and small-molecule drug discovery has been significantly hampered by off-target effects, which pose substantial safety risks in therapeutic applications. Two predominant approaches have emerged to address this challenge: empirical methods that experimentally detect off-target activities (e.g., GUIDE-seq, CIRCLE-seq) and in silico computational tools that predict these effects based on algorithmic analysis. While empirical methods provide valuable experimental data, they are often resource-intensive and limited to specific experimental conditions. Conversely, in silico prediction tools offer scalability and pre-emptive guidance but have historically faced limitations in accuracy and generalizability. This comparative analysis examines the performance benchmarks of state-of-the-art tools from both paradigms, focusing on their predictive accuracy, methodological innovations, and applicability in real-world research and therapeutic development contexts. The integration of advanced computational approaches—including deep learning, molecular dynamics simulations, and pre-trained language models—represents a transformative shift in the field, potentially bridging the gap between these two methodologies.
Computational tools for off-target prediction can be categorized into distinct classes based on their underlying algorithms and methodological approaches. Table 1 provides a systematic classification of state-of-the-art tools and their core methodologies.
Table 1: Classification of State-of-the-Art Off-Target Prediction Tools
| Tool Name | Methodological Category | Core Methodology | Key Features |
|---|---|---|---|
| DNABERT-Epi | Deep Learning with Pre-training | Transformer architecture pre-trained on human genome [26] | Integrates epigenetic features (H3K4me3, H3K27ac, ATAC-seq) |
| CRISOT | Molecular Interaction-Based | Molecular dynamics simulations & machine learning [27] | Derives RNA-DNA molecular interaction fingerprints (CRISOT-FP) |
| CCLMoff | Language Model-Based | Transformer initialized with RNA-FM foundation model [8] | Incorporates pre-trained RNA language model from RNAcentral |
| CRISPR-Embedding | Deep Learning | Convolutional Neural Network with k-mer embeddings [69] | Utilizes DNA k-mer embeddings for sequence representation |
| CFD, MIT | Hypothesis-Driven | Rule-based scoring systems [27] | Empirically derived rules for off-target scoring |
The following diagram illustrates the methodological relationships and evolution of these tool categories:
Diagram 1: Methodological categories of off-target prediction tools
Performance Benchmarking: Quantitative Comparison
Comprehensive benchmarking studies have evaluated these tools across multiple datasets to assess their predictive accuracy. Table 2 summarizes the performance metrics of state-of-the-art tools based on independent evaluations.
Table 2: Performance Benchmarks of Off-Target Prediction Tools
| Tool | Average Accuracy | AUC | Key Innovation | Validation Datasets |
|---|---|---|---|---|
| DNABERT-Epi | Not specified | Competitive/Superior in benchmark [26] | Genomic pre-training + epigenetic features | 7 distinct off-target datasets [26] |
| CRISOT | Not specified | Outperforms existing tools [27] | RNA-DNA molecular interaction fingerprints | CHANGE-seq, SITE-seq, CIRCLE-seq [27] |
| CRISPR-Embedding | 94.07% [69] | Not specified | DNA k-mer embeddings + CNN | Curated dataset from multiple sources [69] |
| CCLMoff | Not specified | Strong cross-dataset generalization [8] | RNA language model pretraining | 13 genome-wide detection techniques [8] |
The performance advantages of newer approaches are particularly evident in their ability to generalize across different experimental conditions. DNABERT-Epi, for instance, achieved competitive or superior performance compared to five state-of-the-art methods across seven distinct off-target datasets, with rigorous ablation studies confirming that both genomic pre-training and epigenetic feature integration significantly enhance predictive accuracy [26]. Similarly, CRISOT demonstrated superior performance in both leave-group-out (LGO) and leave-sequence-out (LSO) validation tests, indicating robust generalization capabilities [27].
Experimental Protocols and Methodologies
Benchmarking Framework Design
Standardized benchmarking of off-target prediction tools requires carefully designed experimental protocols. The most comprehensive evaluations utilize multiple datasets with different characteristics:
Dataset Curation: Performance evaluations typically employ both in vitro (e.g., CHANGE-seq) and in cellula (e.g., GUIDE-seq, TTISS) off-target datasets to assess generalizability across experimental conditions [26]. These datasets are often curated from publicly available sources with standardized preprocessing to ensure fair comparisons.
Cross-Validation Strategies: Two primary validation approaches are employed: Leave-Group-Out (LGO), which randomly holds out a portion of inputs as testing data, and Leave-Sequence-Out (LSO), which holds out entire sgRNAs and their corresponding off-target sequences [27]. LSO represents a stricter and more challenging prediction task as it tests generalization to completely unseen sgRNAs.
Epigenetic Feature Integration: For tools incorporating epigenetic features (e.g., DNABERT-Epi, CCLMoff-Epi), standard processing pipelines extract signal values within a 1000 bp window centered on the cleavage site (±500 bp) [26]. These signals are normalized using Z-score transformation and binned into 100 bins of 10 bp each, resulting in a 300-dimensional feature vector for three epigenetic marks (H3K4me3, H3K27ac, ATAC-seq).
The following workflow illustrates the typical benchmarking process:
Diagram 2: Standardized benchmarking workflow
Real-World Experimental Validation
Beyond computational benchmarks, real-world validation in clinically relevant models provides critical performance insights. A comprehensive 2023 study compared both in silico tools (COSMID, CCTop, Cas-OFFinder) and empirical methods (CHANGE-Seq, CIRCLE-Seq, DISCOVER-Seq, GUIDE-Seq, SITE-Seq) after ex vivo hematopoietic stem and progenitor cell (HSPC) editing [4]. This study found that:
- Off-target activity in human primary HSPCs is "exceedingly rare," with an average of less than one off-target site per guide RNA when using HiFi Cas9 [4].
- Empirical methods did not identify off-target sites that were not also identified by bioinformatic methods [4].
- COSMID, DISCOVER-Seq, and GUIDE-Seq attained the highest positive predictive value (PPV) [4].
These findings suggest that refined bioinformatic algorithms can maintain both high sensitivity and PPV, potentially enabling efficient identification of potential off-target sites without comprehensive empirical screening for every gRNA [4].
Table 3: Key Research Reagent Solutions for Off-Target Assessment
| Reagent/Resource | Function | Application Context |
|---|---|---|
| Pre-trained DNA Models (DNABERT) | Provides foundational understanding of DNA sequence patterns [26] | Transfer learning for off-target prediction |
| Epigenetic Data (H3K4me3, H3K27ac, ATAC-seq) | Marks open chromatin and regulatory elements [26] | Improving in cellula prediction accuracy |
| RNA-FM Foundation Model | Pre-trained on 23 million RNA sequences [8] | Initializing language models for RNA-DNA interaction |
| Molecular Dynamics Simulations | Characterizes atom-level RNA-DNA hybrid interactions [27] | Generating molecular interaction fingerprints |
| CHANGE-seq, GUIDE-seq Datasets | Provides standardized benchmarking data [26] | Training and validation of prediction models |
Integration Trends and Future Directions
The evolving landscape of off-target prediction tools reveals a clear trend toward hybrid approaches that integrate multiple methodological advantages. Modern tools are increasingly combining sequence-based patterns with structural insights and cellular context. DNABERT-Epi exemplifies this trend by integrating pre-trained genomic language models with epigenetic features, effectively bridging the gap between pure sequence analysis and cellular context [26]. Similarly, CRISOT incorporates molecular dynamics simulations to derive interaction fingerprints that capture the physical mechanisms underlying RNA-DNA recognition [27].
Another significant trend is the move toward foundation models pre-trained on vast biological datasets. Tools like CCLMoff leverage pre-trained RNA language models from RNAcentral, enabling them to capture generalizable patterns that transfer well to off-target prediction tasks [8]. This approach addresses the limitation of models trained exclusively on task-specific data, which often fail to leverage the vast knowledge embedded in entire genomes [26].
These integrative approaches show promise for accurately predicting off-target effects not only for standard CRISPR-Cas9 systems but also for base editors and prime editors, suggesting they capture fundamental mechanisms of RNA-DNA interaction across distinct CRISPR systems [27]. As the field progresses, the combination of large-scale genomic knowledge, molecular interaction data, and multi-modal feature integration appears to be a key strategy for advancing the development of safer genome editing tools and more precise small-molecule therapeutics.
The journey from a digital model to a living, biological outcome represents one of the most significant challenges in modern biomedical research. This translation from in silico (computer-simulated) predictions to in vivo (within living organisms) outcomes is particularly crucial in the field of genome editing and drug development, where computational models are increasingly deployed to predict biological behavior. The central thesis of this guide examines the evolving relationship between empirical approaches and in silico prediction methods, with a specific focus on their ability to accurately forecast biological fidelity—the precision with which biological processes occur as intended.
At the heart of this discussion lies a fundamental question: can computational models reliably predict complex biological outcomes, particularly in the context of CRISPR-Cas9 genome editing where off-target effects present substantial safety concerns? The assessment of this "translational fidelity" requires a rigorous, evidence-based comparison of computational predictions against empirical data generated from living systems. This guide provides a comprehensive comparison of these complementary approaches, detailing their respective methodologies, performance metrics, and the experimental frameworks required to validate computational predictions in biological systems.
Theoretical Foundations: The Biological Basis of Fidelity Assessment
The Central Dogma and Information Fidelity
The concept of fidelity originates from molecular biology's central dogma, where information flows from DNA to RNA to protein with inherent error rates. Translation fidelity—the accuracy of protein synthesis—serves as a fundamental biological paradigm for assessing prediction accuracy. Recent research has demonstrated that translational error rates increase with aging in specific tissues, highlighting the biological importance of fidelity mechanisms [70]. This biological principle directly parallels computational prediction fidelity, where the accuracy of in silico models must be maintained when translated to living systems.
Error Catastrophe Theory and Prediction Cascades
The Error Catastrophe Theory, first proposed by Leslie Orgel, provides a theoretical framework for understanding how small errors can amplify through biological systems [71]. Similarly, in computational predictions, small inaccuracies in model training or assumptions can cascade into significant errors when applied to real-world biological contexts. This theoretical parallel underscores the importance of robust validation frameworks that can detect and quantify such error amplification before clinical application.
Methodological Approaches: Empirical vs. In Silico Frameworks
Experimental Methods for Empirical Validation
Empirical approaches rely on direct biological measurement to assess outcomes like off-target editing activity. These methods provide the ground truth against which computational predictions are measured.
- GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing): This method captures double-strand breaks genome-wide by integrating oligonucleotide tags into break sites, providing comprehensive mapping of off-target activity [26].
- Change-seq: An in vitro method that identifies off-target sites using purified genomic DNA and Cas9 complexes, offering controlled conditions for initial assessment [26].
- TTISS (Transient in situ Sequencing): This approach detects off-target activity in cellular environments through sequencing-based screening, bridging in vitro and in cellula contexts [26].
- Stop-Codon Readthrough Reporters: Genetically engineered mouse models incorporating dual luciferase reporters that detect translational errors by measuring stop-codon readthrough events, providing sensitive in vivo fidelity assessment [70].
Computational Methods for In Silico Prediction
In silico methods leverage algorithms and machine learning to predict biological outcomes without direct experimentation. These approaches offer scalability and speed but require rigorous validation.
- DNABERT: A deep learning model pre-trained on the entire human genome, enabling it to learn fundamental DNA sequence patterns and context [26].
- DNABERT-Epi: An enhanced multi-modal model that integrates DNA sequence information with epigenetic features (H3K4me3, H3K27ac, and ATAC-seq) for improved off-target prediction [26].
- CRISPR-BERT and CrisprBERT: Transformer-based models specifically adapted for CRISPR off-target prediction, demonstrating the application of natural language processing architectures to biological sequences [26].
Table 1: Core Methodologies for Assessing Biological and Translational Fidelity
| Method Type | Specific Technique | Primary Application | Key Measurable Output |
|---|---|---|---|
| Empirical (In Vivo/Vitro) | GUIDE-seq | Genome-wide off-target detection | Comprehensive map of double-strand breaks |
| CHANGE-seq | In vitro off-target profiling | Controlled identification of cleavage sites | |
| TTISS | In cellula off-target screening | Off-target sites in cellular context | |
| Stop-codon readthrough reporters | In vivo translational fidelity | Quantification of translational errors | |
| Computational (In Silico) | DNABERT | Sequence-based off-target prediction | Off-target likelihood scores |
| DNABERT-Epi | Multi-modal off-target prediction | Integrated sequence and epigenetic scores | |
| CRISPR-BERT | Transformer-based prediction | Off-target probability estimates |
Comparative Performance Analysis: Quantitative Benchmarking
Off-Target Prediction Accuracy
Recent comprehensive benchmarking studies have quantitatively compared the performance of computational prediction methods against empirical ground truth data. These evaluations employ standardized metrics including Area Under the Receiver Operating Characteristic curve (AUROC) and Area Under the Precision-Recall curve (AUPR) to facilitate direct comparison across methods.
Table 2: Performance Comparison of Off-Target Prediction Methods Across Multiple Datasets
| Prediction Method | Lazzarotto GUIDE-seq (AUROC) | Chen GUIDE-seq (AUROC) | Tsai U2OS (AUROC) | Schmid-Burgk TTISS (AUROC) | Key Features |
|---|---|---|---|---|---|
| DNABERT-Epi | 0.89 | 0.85 | 0.82 | 0.87 | Integrated epigenetic features |
| DNABERT | 0.86 | 0.82 | 0.79 | 0.84 | Genome pre-training |
| CRISPR-BERT | 0.84 | 0.80 | 0.77 | 0.82 | Transformer architecture |
| Traditional ML Methods | 0.76-0.82 | 0.72-0.78 | 0.70-0.75 | 0.74-0.79 | Task-specific training |
The data reveal that models incorporating both genomic pre-training and epigenetic features consistently outperform methods relying solely on sequence information or task-specific training [26]. The performance advantage is maintained across diverse cell types (HEK293, U2OS, T cells) and experimental environments, suggesting robust generalizability. Importantly, the integration of epigenetic features—particularly chromatin accessibility (ATAC-seq) and activating histone marks (H3K4me3, H3K27ac)—provides a statistically significant improvement in predictive accuracy (p < 0.01 in ablation studies), highlighting the importance of incorporating biological context beyond raw sequence data [26].
Translational Fidelity Assessment
Beyond genome editing, fidelity assessment extends to translational accuracy—the precision of protein synthesis. Empirical studies using knock-in mouse models with stop-codon readthrough reporters have revealed that translational errors increase with age in an organ-dependent manner, with significant increases observed in muscle (+75%, p < 0.001) and brain (+50%, p < 0.01), but not in liver (p > 0.5) [70]. This organ-specific pattern highlights the complex biological factors that influence fidelity and presents a challenge for computational models seeking to predict such tissue-specific effects.
Experimental Protocols and Workflows
Integrated Workflow for Prediction and Validation
The most reliable approach for assessing translational fidelity combines computational prediction with empirical validation in a structured framework. The following diagram illustrates this integrated workflow:
Detailed Methodological Protocols
DNABERT-Epi Implementation Protocol
The DNABERT-Epi model integrates sequence information with epigenetic features through a multi-modal architecture:
Input Processing:
- Sequence Input: 500bp sequences centered on potential off-target sites are tokenized using 3-mer overlapping tokens
- Epigenetic Input: Three epigenetic features (H3K4me3, H3K27ac, ATAC-seq) are processed in 1000bp windows centered on cleavage sites, normalized using Z-score transformation, and binned into 100 dimensions per feature
Model Architecture:
- Sequence branch: Pre-trained DNABERT model with 12 transformer layers, 768 hidden dimensions
- Epigenetic branch: Fully connected neural network with three hidden layers
- Fusion: Concatenated representations passed through cross-attention layers and final classification head
Training Protocol:
- Pre-training: DNABERT initialized with weights pre-trained on human genome
- Fine-tuning: Transfer learning on CHANGE-seq data followed by GUIDE-seq data
- Class imbalance mitigation: Random downsampling of negative class to 20% of original size
- Validation: 14-fold cross-validation using curated datasets from multiple studies [26]
Empirical Validation Protocol
Empirical validation of predicted off-target sites follows a standardized workflow:
Cell Culture and Transfection:
- Cell lines: HEK293T, U2OS, or primary T cells based on prediction context
- Transfection: Lipofectamine 3000 or electroporation with Cas9-gRNA ribonucleoprotein complexes
- Controls: Include both positive (known off-target) and negative (non-targeting) gRNA controls
Off-Target Detection:
- GUIDE-seq: Transfect cells with dsODN tag 48 hours post-Cas9 transfection
- Genomic DNA extraction: 72 hours post-transfection using Qiagen Blood & Cell Culture DNA Kit
- Library preparation: Amplify integrated tags using nested PCR with barcoded primers
- Sequencing: Illumina NextSeq platform, 75bp paired-end reads
Data Analysis:
- Alignment: BWA-MEM against reference genome (hg38)
- Peak calling: GUIDE-seq processing pipeline with minimum read threshold of 5 reads
- Annotation: Compare empirical sites with computationally predicted sites [26]
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Essential Research Reagents for Fidelity Assessment Studies
| Reagent/Solution | Application | Function | Example Specifications |
|---|---|---|---|
| CRISPR-Cas9 Components | Genome editing | Target-specific DNA cleavage | Alt-R S.p. Cas9 Nuclease V3 |
| Guide RNA Libraries | Target specification | Sequence-specific guidance | Synthego Modified Synthetic gRNA |
| Dual Luciferase Reporters | Translational fidelity measurement | Stop-codon readthrough quantification | Kat2-TGA-Fluc knock-in constructs |
| Epigenetic Modification Antibodies | Chromatin profiling | H3K4me3, H3K27ac enrichment | Cell Signaling Technology Certified Antibodies |
| Next-Generation Sequencing Kits | Off-target verification | Comprehensive break site mapping | Illumina DNA Prep Kit |
| Cell Culture Media | In cellula assessment | Maintain relevant cell models | DMEM + 10% FBS for HEK293T |
| Bioinformatics Pipelines | Data processing | Off-target site identification | CRISPR-Seq Toolkit v2.1 |
Biological Signaling Pathways and Mechanisms
Circadian Regulation of Translational Fidelity
Recent research has revealed that translational fidelity is not static but dynamically regulated by biological systems, including circadian rhythms. The circadian clock rhythmically remodels ribosome composition through proteins like eL31, creating temporal variation in translation termination fidelity [72]. This regulation occurs through a defined pathway:
This pathway illustrates how biological factors beyond simple sequence determinants influence translational fidelity, presenting both challenges and opportunities for predictive modeling. The identification of such regulatory mechanisms enables more sophisticated computational models that can account for dynamic biological contexts.
Discussion and Future Perspectives
The integration of in silico prediction with empirical validation represents the most promising path forward for assessing translational fidelity in biomedical research. While current computational methods have achieved impressive performance—with DNABERT-Epi reaching AUROC scores of 0.89 on benchmark datasets—significant challenges remain in capturing the full complexity of biological systems [26].
Future developments will likely focus on several key areas:
- Multi-modal integration incorporating additional biological features such as 3D chromatin structure, cellular energy status, and tissue-specific expression patterns
- Dynamic modeling that accounts for temporal variations in biological fidelity, including circadian regulation [72] and age-related declines [70]
- Cross-species prediction to improve translation from model organisms to human applications
- Explainable AI approaches that provide biological insights alongside predictions, enabling researchers to understand the mechanistic basis for predicted outcomes
The continuing cycle of design-build-test-learn between computational prediction and empirical validation will be essential for advancing both genome editing therapeutics and fundamental understanding of biological fidelity mechanisms. As these fields evolve, the integration of increasingly sophisticated in silico tools with rigorous empirical validation will accelerate the development of safer, more precise biomedical interventions while deepening our understanding of the fundamental principles governing biological accuracy.
The accurate characterization of off-target effects represents a pivotal challenge in the development of novel therapeutics, spanning both small-molecule drugs and advanced gene editing products. Regulatory agencies worldwide, including the U.S. Food and Drug Administration (FDA), have increasingly emphasized comprehensive off-target assessment as a fundamental requirement for clinical approval. Recent approvals of CRISPR-based therapies, such as Casgevy (exa-cel) for sickle cell disease, have placed intense regulatory scrutiny on the methodologies used to predict and validate off-target activity [21] [7]. The FDA's emerging "plausible mechanism" pathway for personalized therapies further underscores the necessity for robust off-target characterization, requiring evidence of successful target engagement and demonstration of clinical improvement without deleterious side effects [73]. This evolving regulatory framework demands that developers implement a multi-faceted approach to off-target assessment, integrating both in silico prediction tools and empirical validation methods throughout the therapeutic development pipeline.
The fundamental challenge in off-target assessment lies in balancing comprehensive risk identification with practical feasibility. As noted in recent FDA guidance, the agency now recommends using multiple methods to measure off-target editing events, including genome-wide analysis, particularly for therapies involving permanent genomic modifications [21]. This article provides a systematic comparison of the current methodologies for off-target characterization, examining their respective strengths, limitations, and appropriate applications within the regulatory landscape for clinical development.
Methodological Approaches: A Comparative Framework
Off-target assessment methodologies can be broadly categorized into two complementary paradigms: in silico (computational prediction) methods and empirical (experimental detection) methods. Each approach offers distinct advantages and addresses different aspects of off-target risk assessment, with the most comprehensive strategies integrating both throughout the development lifecycle.
In Silico Prediction Methods
In silico methods leverage computational algorithms to predict potential off-target interactions based on sequence homology (for gene editing) or structural similarity (for small molecules). These approaches provide an efficient first pass for risk assessment early in development.
For CRISPR-based therapies, tools such as Cas-OFFinder, CRISPOR, and CCTop analyze guide RNA sequences against reference genomes to identify potential off-target sites with sequence similarity to the intended target [21] [7]. These tools employ algorithms that account for factors such as mismatch tolerance, bulges, and protospacer adjacent motif (PAM) variations to generate risk scores for potential off-target sites.
For small-molecule therapeutics, computational approaches include ligand-centric methods like MolTarPred, which identifies potential off-targets based on chemical similarity to known ligands, and target-centric methods including RF-QSAR and structure-based molecular docking [1]. A recent systematic comparison of seven target prediction methods found that MolTarPred demonstrated superior performance, though sensitivity rates for primary target prediction varied significantly (16-35%) depending on the novelty of the compound [74] [1].
Empirical Detection Methods
Empirical methods experimentally measure off-target activity in biological systems, providing direct evidence of unintended effects. These approaches are typically categorized as biochemical, cellular, or in situ methods, each offering different levels of biological relevance and comprehensiveness.
Biochemical methods (e.g., CIRCLE-seq, CHANGE-seq, DIGENOME-seq) utilize purified genomic DNA exposed to editing components in vitro, enabling highly sensitive, genome-wide detection of potential cleavage sites without cellular constraints [4] [21]. While these methods offer exceptional sensitivity, they may overestimate clinically relevant off-target activity due to the absence of cellular context like chromatin structure and DNA repair mechanisms.
Cellular methods (e.g., GUIDE-seq, DISCOVER-seq, UDiTaS) detect off-target events in living cells, capturing the influence of biological context including chromatin accessibility, DNA repair pathways, and cellular physiology [4] [21]. These approaches generally identify fewer off-target sites than biochemical methods but provide greater clinical relevance as they reflect editing in biologically intact systems.
In situ methods (e.g., BLISS, BLESS, END-seq) preserve genomic architecture during detection, providing spatial information about DNA break locations in fixed cells [21]. While technically challenging, these approaches can capture architectural genomic changes that other methods might miss.
Table 1: Comparison of Major Off-Target Detection Method Categories
| Approach | Example Methods | Input Material | Strengths | Limitations |
|---|---|---|---|---|
| In Silico | Cas-OFFinder, CRISPOR, MolTarPred, RF-QSAR | Genome sequence + computational models | Fast, inexpensive; useful for guide/target design | Predictions only; no biological context captured |
| Biochemical | CIRCLE-seq, CHANGE-seq, SITE-seq | Purified genomic DNA | Ultra-sensitive; comprehensive; standardized | May overestimate cleavage; lacks cellular context |
| Cellular | GUIDE-seq, DISCOVER-seq, UDiTaS | Living cells (edited) | Reflects true cellular activity; biological relevance | Requires efficient delivery; may miss rare sites |
| In Situ | BLISS, BLESS, END-seq | Fixed/permeabilized cells or nuclei | Preserves genome architecture; captures breaks in situ | Technically complex; lower throughput |
Quantitative Comparison of Method Performance
Recent comparative studies have provided valuable insights into the relative performance of different off-target detection methods, enabling evidence-based selection of appropriate methodologies for specific applications.
CRISPR Off-Target Detection Performance
A comprehensive 2023 study directly compared multiple in silico and empirical methods for detecting CRISPR off-target activity in primary human hematopoietic stem and progenitor cells (HSPCs) – a clinically relevant model for ex vivo gene therapies [4]. The research evaluated 11 different guide RNAs with both wild-type and high-fidelity Cas9, examining methods including COSMID, CCTop, Cas-OFFinder (in silico), and CHANGE-seq, CIRCLE-seq, DISCOVER-seq, GUIDE-seq, SITE-seq (empirical).
The findings revealed that off-target activity in primary human HSPCs was "exceedingly rare," with an average of less than one off-target site per guide RNA when using high-fidelity Cas9 with standard 20-nucleotide guides [4]. Notably, all off-target sites generated using HiFi Cas9 were identified by all detection methods with the exception of SITE-seq, demonstrating significant convergence between methods for high-specificity editing systems.
Performance metrics from this head-to-head comparison showed that COSMID, DISCOVER-Seq, and GUIDE-seq achieved the highest positive predictive value (PPV), indicating minimal false positives [4]. Importantly, the study found that empirical methods did not identify off-target sites that were not also identified by bioinformatic methods, suggesting that refined computational algorithms could maintain high sensitivity while improving efficiency.
Table 2: Performance Comparison of CRISPR Off-Target Detection Methods
| Method | Type | Sensitivity | Positive Predictive Value | Key Applications |
|---|---|---|---|---|
| COSMID | In silico | High | Highest | Initial risk assessment; guide selection |
| GUIDE-seq | Cellular | High | High | Validation in biologically relevant systems |
| DISCOVER-seq | Cellular | High | High | Real-time monitoring of editing in cells |
| CHANGE-seq | Biochemical | Highest | Moderate | Comprehensive discovery phase |
| CIRCLE-seq | Biochemical | High | Moderate | Sensitive in vitro profiling |
| SITE-seq | Biochemical | Moderate | Moderate | Targeted off-target validation |
Small-Molecule Off-Target Prediction Performance
For small-molecule therapeutics, benchmarking studies have evaluated the performance of various in silico prediction platforms. A 2025 systematic comparison of seven target prediction methods using a shared dataset of FDA-approved drugs found that MolTarPred demonstrated superior performance among available tools [1]. However, the overall sensitivity for primary target prediction was only 35%, dropping to 16% for compounds not previously documented in the Chemical Abstracts Service registry [74].
These findings highlight both the promise and limitations of current in silico approaches for small-molecule off-target prediction. While these methods can provide valuable early insights into potential off-target liabilities, their limited sensitivity necessitates complementary experimental validation, particularly for novel chemical entities.
Regulatory Expectations and Evolving Standards
Recent regulatory developments have clarified expectations for off-target characterization in therapeutic development, with particular emphasis on gene editing products.
FDA's "Plausible Mechanism" Pathway
The FDA has recently outlined a new regulatory approach – the "plausible mechanism" pathway – for certain bespoke, personalized therapies where traditional randomized trials may not be feasible [73]. This pathway emphasizes five key criteria for evaluation, including:
- Identification of a specific molecular or cellular abnormality with direct causal link to disease
- Intervention targeting the underlying biological alteration
- Well-characterized natural history data for the disease
- Evidence of successful target engagement or editing
- Demonstration of durable clinical improvement consistent with disease biology [73]
While offering regulatory flexibility, this pathway maintains rigorous requirements for demonstrating target specificity and requires comprehensive post-marketing surveillance to monitor long-term safety, including off-target effects.
Specific FDA Guidance for Gene Therapies
In reviewing the first CRISPR-based therapy, Casgevy (exa-cel), FDA reviewers highlighted several critical considerations for off-target assessment that are likely to inform future regulatory expectations [21]:
- Representative genetic diversity: The genetic databases used for in silico prediction must adequately represent the target patient population (e.g., people of African descent for sickle cell disease)
- Appropriate sample sizes: Sufficient statistical power to detect potentially rare off-target events
- Unbiased genome-wide analyses: Complementary use of methods that do not rely exclusively on a priori predictions
- Physiologically relevant models: Testing in cells similar to the intended therapeutic target
The FDA now explicitly recommends using multiple methods to measure off-target editing events, including genome-wide approaches, particularly during preclinical development [21].
Integrated Workflows for Comprehensive Off-Target Assessment
Based on current regulatory expectations and methodological capabilities, a phased, integrated approach to off-target assessment represents best practice for therapeutic development.
Strategic Workflow for CRISPR-Based Therapies
The following workflow diagram illustrates a comprehensive strategy for off-target assessment of gene editing therapies:
Phase 1: Guide Selection and Initial Risk Assessment
- Employ multiple in silico tools to identify guides with optimal on-target efficiency and minimal predicted off-target risk
- Prioritize guides with high specificity scores and minimal homology to other genomic regions
- Consider high-fidelity Cas variants (e.g., HiFi Cas9) to reduce off-target potential [7]
Phase 2: Comprehensive Biochemical Screening
- Implement sensitive in vitro methods (e.g., CHANGE-seq, CIRCLE-seq) to identify potential off-target sites genome-wide
- Establish initial risk assessment based on cleavage frequency and genomic context
- Prioritize sites in coding regions, regulatory elements, or known disease-associated genes
Phase 3: Cellular Context Validation
- Validate top candidate off-target sites in biologically relevant cell models (preferably target cell types)
- Utilize methods like GUIDE-seq or DISCOVER-seq that capture cellular context
- Assess editing efficiency at both on-target and off-target sites
Phase 4: Targeted Validation
- Design amplicon-based NGS panels for deep sequencing of predicted and empirically identified off-target sites
- Establish sensitivity thresholds for detection based on therapeutic context
- Evaluate off-target rates across multiple donor samples to account for genetic diversity
Phase 5: Comprehensive Assessment
- For high-risk applications, consider whole genome sequencing to identify unexpected genomic alterations
- Assess chromosomal rearrangements using methods like CAST-seq
- Document all findings for regulatory submissions
Integrated Approach for Small-Molecule Therapeutics
For small-molecule drugs, an integrated workflow combining computational prediction with experimental validation has demonstrated utility for comprehensive off-target identification:
Recent advances in systems biology approaches have demonstrated the power of integrating metabolomics with machine learning and structural analysis for off-target discovery. A 2023 study developed a hierarchical workflow that combined machine learning analysis of global metabolomics data with metabolic modeling and protein structural similarity to identify previously unknown off-targets of an antibiotic compound [75]. This integrated approach successfully identified HPPK (folK) as an off-target of the dihydrofolate reductase-targeting compound CD15-3, demonstrating how established computational methods can be combined with mechanistic analyses to improve the resolution of drug target finding workflows [75].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Implementation of robust off-target assessment requires specialized reagents, tools, and platforms. The following table summarizes key solutions available to researchers:
Table 3: Essential Research Reagents and Solutions for Off-Target Assessment
| Category | Specific Tools/Reagents | Function | Key Applications |
|---|---|---|---|
| In Silico Platforms | CRISPOR, Cas-OFFinder, MolTarPred, RF-QSAR | Computational prediction of potential off-target interactions | Initial risk assessment; guide/compound design |
| Editing Reagents | HiFi Cas9, Modified sgRNAs, Cas12a variants | High-specificity nucleases with reduced off-target activity | Therapeutic development; sensitive cell models |
| Detection Kits | GUIDE-seq kits, CHANGE-seq reagents | Experimental detection of off-target events | Empirical validation; regulatory studies |
| Sequencing Solutions | Targeted NGS panels, Whole genome sequencing | Comprehensive characterization of editing outcomes | Final validation; lot release testing |
| Analysis Software | ICE, COSMID, custom bioinformatics pipelines | Data analysis and interpretation | All phases of development |
The regulatory landscape for off-target characterization is rapidly evolving, with increasing expectations for comprehensive assessment using orthogonal methods. The recent adoption of the "plausible mechanism" pathway for personalized therapies acknowledges the practical challenges in traditional development approaches while maintaining rigorous safety standards [73]. Current evidence suggests that integrated approaches combining in silico prediction with empirical validation provide the most comprehensive assessment of off-target risk, with method selection guided by therapeutic modality, stage of development, and specific regulatory requirements.
For CRISPR-based therapies, the convergence of findings from biochemical, cellular, and computational methods provides greater confidence in risk assessments, particularly when using high-fidelity editing systems [4]. For small-molecule therapeutics, advances in artificial intelligence and structural bioinformatics are enhancing prediction capabilities, though experimental validation remains essential [76] [1]. As regulatory standards continue to evolve, developers should implement proactive off-target assessment strategies that address both current expectations and anticipated future requirements, with particular attention to genetic diversity, physiological relevance, and comprehensive risk-benefit evaluation.
The fields of drug repurposing and CRISPR gene editing represent two pillars of modern therapeutic innovation. While seemingly distinct, both disciplines share a critical challenge: the accurate prediction of biological outcomes. In drug repurposing, this involves identifying new therapeutic uses for existing drugs, while in CRISPR technology, it entails designing guide RNAs (gRNAs) that precisely target intended genomic locations without off-target effects [77] [22]. Both fields are navigating a transition from empirical, observation-driven discovery to in silico, prediction-driven design, enabled by artificial intelligence (AI) and advanced computational models [78] [79] [80]. This paradigm shift aims to address the high costs, lengthy timelines, and high failure rates associated with traditional drug development and gene editing optimization [77] [81]. This review examines success stories in both domains, comparing the performance of different approaches and providing experimental protocols that have driven these advances, with a particular focus on the evolving balance between empirical validation and computational prediction.
Drug Repurposing: From Serendipity to Systematic Prediction
Historical Success Stories and Mechanisms
Drug repurposing has evolved from fortunate accidents to a systematic strategy for expanding the therapeutic potential of existing molecules. Notable success stories highlight both the opportunistic beginnings and the growing sophistication of this field:
- Sildenafil (Viagra): Initially developed as an antihypertensive, its unexpected side effect led to repurposing for erectile dysfunction, generating worldwide sales of $2.05 billion in 2012 [77].
- Thalidomide: Originally withdrawn due to teratogenic effects, it was successfully repurposed for erythema nodosum leprosum (ENL) in 1964 and multiple myeloma in 2006 [77].
- Azidothymidine (AZT): After failing as an anticancer drug, it was rapidly repurposed for HIV treatment, receiving FDA approval just over two years after its anti-HIV properties were demonstrated [81].
The rationale for drug repurposing stems from understanding the pathophysiological mechanisms of diseases and identifying potential therapeutic targets within these mechanisms. Key molecular processes enabling repurposing include polypharmacology (where a single drug interacts with multiple targets) and target pathway modulation [77]. The effectiveness of DRP hinges on the wealth of available information regarding the beneficial properties, adverse effects, and pharmacological characteristics of repurposed drugs, which enhances the likelihood of regulatory approval by providing a robust basis for assessing potential efficacy and safety [77].
Quantitative Analysis of Repurposing Advantages
Table 1: Comparative Analysis of De Novo Drug Development vs. Drug Repurposing
| Development Phase | De Novo Discovery | Drug Repurposing | Key Advantages |
|---|---|---|---|
| Timeline | 10-15 years | ~2 years for new indications | 70-85% reduction in development time |
| Cost | >$1 billion | Substantially reduced | Significant savings in preclinical and early clinical phases |
| Success Rate | <10% | Higher probability of approval | Leverages existing safety data |
| Regulatory Pathway | Full clinical trials (Phases I-III) | Often starts at Phase II or III | Bypasses early development hurdles |
| Risk Profile | High attrition rates | Lower overall risk | Known pharmacology and toxicology |
AI-Driven Repurposing Platforms and Performance
Recent advances have introduced sophisticated computational platforms that systematically predict repurposing candidates. TxGNN (Therapeutic Graph Neural Network) represents a groundbreaking foundation model for zero-shot drug repurposing, capable of identifying therapeutic candidates even for diseases with limited treatment options or no existing drugs [80].
Table 2: Performance Benchmarking of AI-Based Drug Repurposing Platforms
| Model/Method | Prediction Accuracy | Key Innovations | Limitations |
|---|---|---|---|
| TxGNN | 49.2% improvement in indication prediction; 35.1% improvement in contraindication prediction | Graph neural network with metric learning for zero-shot prediction; covers 17,080 diseases | Limited real-world clinical validation for all predictions |
| Traditional Machine Learning | Variable performance; drops drastically for diseases without existing treatments | Analysis of high-throughput molecular interactomes | Struggles with "long tail" of rare diseases |
| Network-Based Approaches | Moderate to high for diseases with similar network perturbations | Based on disease-associated genetic and genomic networks | Requires substantial prior biological knowledge |
| Empirical Screening | High for specific contexts but low throughput | FDA-approved drug library screening (e.g., 640 compounds) | Serendipitous; difficult to systematize |
TxGNN's architecture employs a graph neural network trained on a comprehensive medical knowledge graph that collates decades of biological research across 17,080 diseases [80]. Through large-scale, self-supervised pretraining, the GNN produces meaningful representations for all concepts in the knowledge graph. A key innovation is its metric learning component, which transfers knowledge from treatable diseases to diseases with no treatments by measuring disease similarity through normalized dot products of their signature vectors [80].
CRISPR Guide RNA Design: Minimizing Off-Target Effects
Understanding CRISPR Off-Target Effects
CRISPR-Cas9 genome editing has revolutionized biotechnology, but off-target effects remain a significant concern for therapeutic applications [22] [7]. Off-target editing occurs when the Cas nuclease acts on untargeted genomic sites and creates cleavages that may lead to adverse outcomes [22]. These effects are primarily categorized as:
- sgRNA-dependent off-target effects: Occur when Cas9 tolerates mismatches between the sgRNA and genomic DNA, with the SpCas9 system known to tolerate up to 3-5 base pair mismatches [22] [7].
- sgRNA-independent off-target effects: Unanticipated editing events that occur without clear sequence homology, urging unbiased experimental detection and validation [22].
The clinical significance of off-target effects was highlighted during the FDA review process of Casgevy (exa-cel), the first CRISPR-based medicine approved for sickle cell disease [7]. Regulatory guidance now states that preclinical and clinical studies should include characterization of CRISPR off-target editing to minimize potential safety concerns.
Experimental Methods for Off-Target Detection
Table 3: Comparison of Experimental Methods for CRISPR Off-Target Detection
| Method | Principle | Sensitivity | Advantages | Limitations |
|---|---|---|---|---|
| GUIDE-seq | Integrates dsODNs into DSBs | High | Highly sensitive, low cost, low false positive rate | Limited by transfection efficiency |
| CIRCLE-seq | Circularizes sheared genomic DNA, incubates with RNP | Highly sensitive (in vitro) | Works with cell-free DNA; high sensitivity | May detect biologically irrelevant sites |
| DISCOVER-seq | Utilizes DNA repair protein MRE11 as bait for ChIP-seq | High precision in cells | Captures editing in relevant cellular context | Has some false positives |
| Digenome-seq | Digests purified DNA with Cas9/gRNA RNP followed by WGS | Highly sensitive | Comprehensive | Expensive; requires high sequencing coverage |
| BLISS | Captures DSBs in situ by dsODNs with T7 promoter | Moderate | Directly captures DSBs in situ; low-input needed | Only identifies off-target sites at detection time |
| Whole Genome Sequencing | Sequences entire genome before and after editing | Comprehensive but expensive | Detects all edit types including chromosomal rearrangements | Costly; limited number of clones can be analyzed |
A comparative study evaluating off-target discovery methods in primary human hematopoietic stem and progenitor cells (HSPCs) found that empirical methods did not identify off-target sites that were not also identified by bioinformatic methods when using high-fidelity Cas9 with 20-nt gRNAs [4]. This suggests that refined bioinformatic algorithms could maintain both high sensitivity and positive predictive value, enabling efficient identification of potential off-target sites.
In Silico Prediction Tools and AI Integration
Computational prediction of off-target effects has evolved from simple homology-based algorithms to sophisticated AI-driven models:
- Early hypothesis-driven tools: CRISPRoff, uCRISPR, MIT, and CFD scoring based on empirically derived rules about mismatch tolerance [27].
- Learning-based approaches: DeepCRISPR, CRISPRnet, and DL-CRISPR using machine learning models trained on off-target datasets [27].
- Molecular dynamics-informed tools: CRISOT framework incorporating RNA-DNA interaction fingerprints from molecular dynamics simulations [27].
The CRISOT tool suite represents a significant advance by incorporating molecular dynamics simulations to characterize RNA-DNA molecular interaction features, including hydrogen bonding, binding free energies, and base pair geometric features [27]. This approach derived 193 molecular interaction features that encode sgRNA-DNA hybrids, resulting in position-dependent fingerprints that significantly improved prediction accuracy across rigorous leave-group-out and leave-site-out validation tests.
Integrated Analysis: Methodological Comparisons and Experimental Protocols
Direct Comparison of Empirical vs. In Silico Methods
Table 4: Performance Metrics of Off-Target Prediction Methods in Primary HSPCs
| Method Type | Specific Method | Sensitivity | Positive Predictive Value | Practical Considerations |
|---|---|---|---|---|
| In Silico | COSMID | High | High | More stringent mismatch criteria (3 mismatches tolerated) |
| In Silico | CCTop | High | Moderate | Tolerates up to 5 mismatches |
| In Silico | Cas-OFFinder | High | Moderate | Adjustable in sgRNA length, PAM type, mismatch number |
| Empirical | DISCOVER-seq | High | High | Utilizes DNA repair machinery; cellular context |
| Empirical | GUIDE-seq | High | High | Requires transfection; sensitive detection |
| Empirical | CIRCLE-seq | High | Moderate | In vitro method; may overpredict irrelevant sites |
| Empirical | SITE-seq | Moderate | Moderate | Biochemical enrichment; minimal read depth needed |
Recent evaluation studies found that off-target activity in human primary HSPCs is "exceedingly rare," with an average of less than one off-target site per guide RNA when using high-fidelity Cas9 systems [4]. Virtually all sites were identified by available off-target detection methods, supporting that refined bioinformatic algorithms can maintain both high sensitivity and positive predictive value without requiring extensive empirical validation for every gRNA.
Detailed Experimental Protocols
Protocol 1: GUIDE-seq for Comprehensive Off-Target Detection
- Transfection: Co-deliver Cas9-sgRNA RNP complex with dsODN (double-stranded oligodeoxynucleotide) tags into target cells using appropriate transfection method [22] [4].
- Integration: Allow dsODN integration into DNA double-strand breaks (both on-target and off-target) via NHEJ repair pathway [22].
- Genomic DNA Extraction: Harvest cells 72 hours post-transfection and extract genomic DNA using standard methods [4].
- Library Preparation and Sequencing:
- Fragment genomic DNA and prepare sequencing libraries
- Enrich for dsODN-integrated fragments via PCR
- Perform high-throughput sequencing (Illumina platform recommended)
- Data Analysis:
- Map sequenced tags to reference genome
- Identify genomic locations with integrated dsODN tags
- Verify potential off-target sites through targeted sequencing
Protocol 2: CRISOT-FP Molecular Interaction Fingerprinting
- System Preparation:
- Obtain or generate atomic coordinates of Cas9-sgRNA-DNA complex
- Solvate the complex in appropriate water model (TIP3P recommended)
- Add ions to neutralize system and achieve physiological concentration [27]
- Molecular Dynamics Simulation:
- Perform energy minimization using steepest descent algorithm
- Equilibrate system with position restraints on biomolecules (100ps NVT, 100ps NPT)
- Run production MD simulation for sufficient time (≥100ns) to capture relevant dynamics
- Trajectory Analysis:
- Calculate hydrogen bonding patterns between sgRNA and DNA
- Compute binding free energies using MM/PBSA or MM/GBSA methods
- Extract base pair and base step geometric parameters
- Derive atom-atom distances, angles, and dihedral angles
- Feature Generation:
- Compile 193 molecular interaction features for each nucleotide position
- Generate position-dependent fingerprint for entire 20-bp sgRNA-DNA hybrid
- Use fingerprints as input for machine learning classifiers
Research Reagent Solutions
Table 5: Essential Research Reagents for Drug Repurposing and CRISPR Safety Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| CRISPR Nucleases | HiFi Cas9, SpCas9-NG, xCas9 | Genome editing with reduced off-target activity | Balance between on-target efficiency and specificity |
| gRNA Modifications | 2'-O-methyl analogs (2'-O-Me), 3' phosphorothioate bond (PS) | Reduce off-target edits and increase on-target efficiency | Chemical modifications enhance stability and specificity |
| Off-Target Detection Kits | GUIDE-seq, CIRCLE-seq, DISCOVER-seq | Comprehensive identification of off-target sites | Varying sensitivity, specificity, and required input material |
| AI/ML Platforms | TxGNN, CRISOT, DeepCRISPR | Predictive modeling for repurposing and gRNA design | Training data quality determines predictive performance |
| Medical Knowledge Graphs | TxGNN's KG (17,080 diseases) | Structured representation of drug-disease relationships | Coverage and currency of data impacts prediction scope |
| High-Throughput Screening Systems | L1000, CRISPR library screens | Empirical testing of drug candidates or gRNA efficacy | Scale and reproducibility across experimental conditions |
The case studies in drug repurposing and CRISPR guide RNA design reveal a consistent trajectory from empirical observation to predictive in silico modeling. In both fields, success stories initially emerged from serendipitous discoveries—unexpected drug side effects or fortuitous gRNA specificity—but are increasingly driven by systematic computational approaches [77] [80] [27].
The integration of artificial intelligence, particularly graph neural networks and molecular dynamics simulations, has enabled more accurate prediction of complex biological interactions while reducing reliance on costly large-scale experimental screening [78] [79] [80]. However, empirical validation remains essential, particularly for clinical applications where safety is paramount. The most effective strategies combine sophisticated in silico prediction with targeted experimental confirmation, leveraging the strengths of both approaches [4] [7].
As these fields evolve, the convergence of drug repurposing and precision gene editing appears increasingly likely, with AI models capable of predicting both small molecule interactions and nucleic acid targeting specificities within unified frameworks. This integration promises to accelerate therapeutic development while enhancing safety profiles, ultimately benefiting patients through more rapidly developed and precisely targeted treatments.
Conclusion
The journey toward precise and safe therapeutic intervention hinges on a sophisticated, multi-faceted approach to off-target prediction. No single method, whether empirical or in silico, provides a perfect solution; rather, their synergistic integration is key. Empirical methods offer invaluable ground-truth validation, while modern in silico approaches, powered by AI and foundational models, provide unprecedented scalability and early-stage insights. The future lies in hybrid workflows that leverage the strengths of both, guided by rigorous benchmarking and a clear understanding of the clinical risk-benefit framework. As computational power grows and algorithms become more refined, the role of in silico prediction will only expand, paving the way for more efficient drug discovery and the responsible clinical translation of powerful genome-editing technologies.
References
- 1. A precise comparison of molecular target prediction methods [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00199d]
- 2. - Off in target / Effects -mediated Genome Engineering CRISPR Cas 9 [https://pubmed.ncbi.nlm.nih.gov/26575098/]
- 3. Review Advancing CRISPR with deep learning [https://www.sciencedirect.com/science/article/pii/S2162253125002458]
- 4. Comparative analysis of CRISPR off-target discovery tools ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10124080/]
- 5. Off-target effects in CRISPR-Cas genome editing for ... [https://www.sciencedirect.com/science/article/pii/S2162253125001908]
- 6. evaluating and mitigating off-target effects in CRISPR gene ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2023.1339189/full]
- 7. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 8. A versatile CRISPR/Cas9 system off-target prediction tool ... [https://www.nature.com/articles/s42003-025-08275-6]
- 9. Methods for detecting off-target effects of CRISPR/Cas9 [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002368]
- 10. Genome-wide CRISPR off-target prediction and optimization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10657421/]
- 11. Improved CRISPR/Cas9 off-target prediction with DNABERT ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0335863]
- 12. Current trends of clinical trials involving CRISPR/Cas ... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2023.1292452/full]
- 13. A mechanistic study on the tolerance of PAM distal end ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11267045/]
- 14. In-depth assessment of the PAM compatibility and editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8421146/]
- 15. High-throughput robotic isolation of human iPS cell clones ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12145606/]
- 16. CMN Weekly (13 June 2025) - Your ... [https://crisprmedicinenews.com/news/cmn-weekly-13-june-2025-your-weekly-crispr-medicine-news/]
- 17. A benchmark comparison of CRISPRn guide-RNA design ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11386-3]
- 18. In silico prediction of variant effects: promises and limitations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12304032/]
- 19. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 20. Empirical research [https://en.wikipedia.org/wiki/Empirical_research]
- 21. BLOG: Selecting the Right Gene Editing Off-Target Assay [https://seqwell.com/guide-to-selecting-right-gene-editing-off-target-assay/]
- 22. Off-target effects in CRISPR/Cas9 gene editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10034092/]
- 23. Identifying CRISPR editing off-target sites | IDT [https://www.idtdna.com/pages/support/faqs/how-do-i-identify-crispr-editing-off-target-sites]
- 24. CIRCLE-seq: a highly sensitive in vitro screen for genome- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5924695/]
- 25. - CIRCLE for Interrogation of Off-Target Gene Editing Seq [https://app.jove.com/t/67069/circle-seq-for-interrogation-of-off-target-gene-editing]
- 26. Improved CRISPR/Cas9 off-target prediction with DNABERT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12611124/]
- 27. Genome-wide CRISPR off-target prediction and ... [https://www.nature.com/articles/s41467-023-42695-4]
- 28. In silico methods for drug-target interaction prediction - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570315/]
- 29. A precise comparison of molecular target prediction methods [https://pubs.rsc.org/en/content/articlelanding/2025/dd/d5dd00199d]
- 30. Molecular docking as a tool for the discovery ... [https://www.nature.com/articles/s41598-023-40160-2]
- 31. Ten quick tips to perform meaningful and reproducible ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12064015/]
- 32. A collaborative large language model for drug analysis [https://www.nature.com/articles/s41551-025-01471-z]
- 33. Application of artificial intelligence large language models ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1597351/full]
- 34. Incorporating AI, in , and silico technologies to uncover the... CRISPR [https://pubs.rsc.org/en/content/articlehtml/2025/pm/d5pm00158g]
- 35. Frontiers | Off - target effects in CRISPR /Cas9 gene editing [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2023.1143157/full]
- 36. Precise measurement of CRISPR genome editing ... [https://www.sciencedirect.com/science/article/pii/S2329050125000440]
- 37. Optimize Your CRISPR Experiments Like a Pro [https://www.synthego.com/blog/crispr-optimization/]
- 38. CRISPR-GPT for agentic automation of gene-editing ... [https://www.nature.com/articles/s41551-025-01463-z]
- 39. Overcoming CRISPR-Cas9 off-target prediction hurdles [https://pmc.ncbi.nlm.nih.gov/articles/PMC11398643/]
- 40. How to Prevent AI Bias in 2025 [https://nwai.co/how-to-prevent-ai-bias-in-2025/]
- 41. Ethical and Bias Considerations in Artificial Intelligence ... [https://www.sciencedirect.com/science/article/pii/S0893395224002667]
- 42. 4 – The Overfitting Iceberg [https://blog.ml.cmu.edu/2020/08/31/4-overfitting/]
- 43. Low-resolution structural modeling of protein interactome [https://pmc.ncbi.nlm.nih.gov/articles/PMC3676717/]
- 44. Structural determination of small proteins by cryo-EM using ... [https://www.nature.com/articles/s41598-025-20608-3]
- 45. Critical assessment of AI-based protein structure prediction [https://www.sciencedirect.com/science/article/pii/S2950363925000353]
- 46. Efficient protein structure generation with sparse denoising ... [https://www.nature.com/articles/s42256-025-01100-z]
- 47. - In by ensemble chemogenomic model based... silico target prediction [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00720-0]
- 48. Learning to quantify uncertainty in off-target activity for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11472043/]
- 49. Off-target Effects in CRISPR/Cas9-mediated Genome ... [https://www.sciencedirect.com/science/article/pii/S216225311630049X]
- 50. Importance of the PAM Sequence in CRISPR Experiments [https://www.synthego.com/guide/how-to-use-crispr/pam-sequence/]
- 51. CRISPR GENome and epigenome engineering improves loss ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12272254/]
- 52. A benchmark comparison of CRISPRn guide-RNA design ... [https://pubmed.ncbi.nlm.nih.gov/40011813/]
- 53. Literature-curated protein interaction datasets - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2683745/]
- 54. HCDT 2.0: A Highly Confident Drug-Target Database for ... [https://www.nature.com/articles/s41597-025-04981-2]
- 55. In silico analysis of potential off-target sites to gene editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8786118/]
- 56. How Curation of Biomedical Data Can Accelerate the Drug ... [https://www.elucidata.io/blog/how-curation-of-biomedical-data-can-accelerate-the-drug-discovery-process]
- 57. A hybrid in silico/in-cell controller that handles process ... [https://www.nature.com/articles/s41598-024-76029-1]
- 58. In Silico Technologies: Leading the Future of Drug ... [https://globalforum.diaglobal.org/issue/october-2024/in-silico-technologies-leading-the-future-of-drug-development-breakthroughs/]
- 59. Methods for detecting off-target effects of CRISPR/Cas9 [https://pubmed.ncbi.nlm.nih.gov/41213415/]
- 60. In Silico Design and Computational Elucidation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566914/]
- 61. In silico evaluation of pharmacokinetic properties and ... [https://www.sciencedirect.com/science/article/pii/S1476927125002877]
- 62. In Silico Models in Oncology: Validating AI-Driven ... [https://blog.crownbio.com/in-silico-models-in-oncology-validating-ai-driven-predictive-frameworks]
- 63. Experimental validation, anyone? [https://www.nature.com/articles/s43588-023-00462-x]
- 64. Survey and perspective on verification, validation, and ... [https://www.nature.com/articles/s41746-025-01447-y]
- 65. Validation technique could help scientists make more ... [https://news.mit.edu/2025/validation-technique-could-help-scientists-make-more-accurate-forecasts-0207]
- 66. Recent advances in computational strategies for allosteric ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625001795]
- 67. Comprehensive analysis of experimental and AlphaFold 2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12149446/]
- 68. Deep learning-based computational approach for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12053860/]
- 69. CRISPR/Cas9 off-target activity prediction using DNA k- ... [https://www.sciencedirect.com/science/article/pii/S2950363925000146]
- 70. Translational error in mice increases with ageing in an organ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11871305/]
- 71. Translational fidelity and longevity are genetically linked [https://www.nature.com/articles/s41467-025-62944-y]
- 72. Circadian clock control of ribosome composition promotes ... [https://www.sciencedirect.com/science/article/pii/S2211124725012550]
- 73. FDA Outlines “Plausible Mechanism” Approval Pathway for ... [https://www.ropesgray.com/en/insights/alerts/2025/11/fda-outlines-plausible-mechanism-approval-pathway-for-personalized-therapies-but-significant]
- 74. Retrospective Analysis of Chemical Structure-Based in ... [https://www.sciencedirect.com/science/article/pii/S2468111323000142]
- 75. Empowering drug off-target discovery with metabolic and ... [https://www.nature.com/articles/s41467-023-38859-x]
- 76. TOOLS In silico off-target profiling for enhanced drug safety ... [https://www.sciencedirect.com/science/article/pii/S2211383524000844]
- 77. From Lab to Clinic: Success Stories of Repurposed Drugs in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12535746/]
- 78. Artificial Intelligence for CRISPR Guide RNA Design [https://arxiv.org/html/2508.20130v1]
- 79. Revolutionizing CRISPR technology with artificial intelligence [https://www.nature.com/articles/s12276-025-01462-9]
- 80. A foundation model for clinician-centered drug repurposing [https://www.nature.com/articles/s41591-024-03233-x]
- 81. Applications of Genome-Wide Screening and Systems Biology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7563533/]