CRISPR-Cas9 Knockout Cell Line Generation: A 2025 Guide from Fundamentals to Clinical Applications
This article provides a comprehensive guide for researchers and drug development professionals on generating CRISPR-Cas9 knockout cell lines.
CRISPR-Cas9 Knockout Cell Line Generation: A 2025 Guide from Fundamentals to Clinical Applications
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on generating CRISPR-Cas9 knockout cell lines. It covers foundational principles, detailed step-by-step protocols, advanced troubleshooting for common challenges like low efficiency and off-target effects, and robust validation strategies. The content synthesizes the latest 2025 methodological advancements, including AI-enhanced sgRNA design and optimized delivery systems, while addressing specific applications in disease modeling, drug discovery, and the development of cell-based potency assays for gene therapies.
CRISPR-Cas9 Knockout Fundamentals: Revolutionizing Genetic Research and Therapeutic Discovery
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) system represents a revolutionary genome editing technology derived from an adaptive immune mechanism in bacteria and archaea [1] [2]. This system provides acquired resistance against invading viruses and plasmids by recognizing and cleaving foreign genetic elements [2]. The technological domestication of this biological system has created a powerful platform for precise genome manipulation across diverse organisms and cell types [3].
Compared to previous genome editing technologies like Zinc Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs), which require complex protein engineering for each new target site, CRISPR-Cas9 offers unprecedented simplicity and programmability [1] [2]. Where ZFNs and TALENs rely on custom-designed protein domains for DNA recognition, the CRISPR-Cas9 system utilizes a simple guide RNA molecule to direct the Cas9 nuclease to specific genomic loci, dramatically reducing the time and expertise required for experimental design [1]. This programmability has positioned CRISPR-Cas9 as the ideal tool for generating knockout cell lines, enabling researchers to investigate gene function with precision and efficiency [3].
Molecular Mechanism of CRISPR-Cas9
Core Components and Their Functions
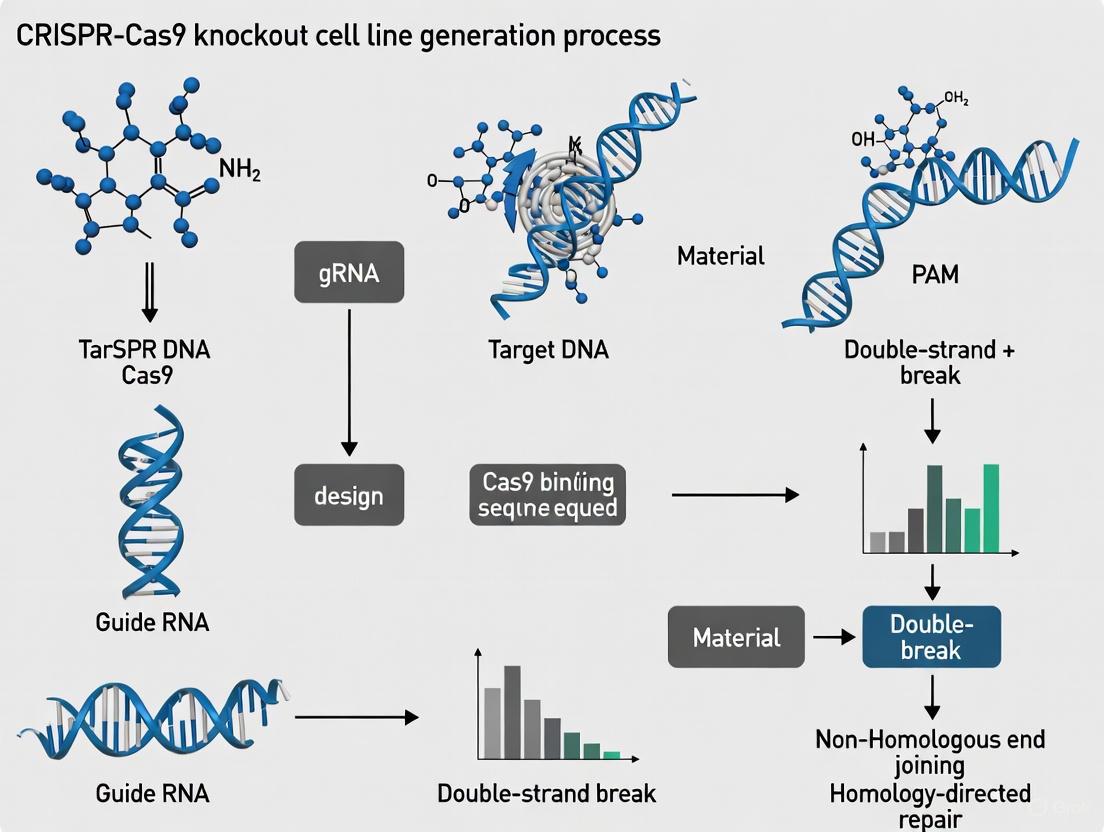
The CRISPR-Cas9 system functions as a two-component complex consisting of the Cas9 nuclease and a guide RNA (gRNA) [1] [2]. The Cas9 protein serves as the effector module that creates double-strand breaks (DSBs) in DNA, while the guide RNA provides the targeting specificity through complementary base pairing [4].
The guide RNA is a synthetic fusion of two natural RNA molecules: the CRISPR RNA (crRNA), which contains the ~20 nucleotide spacer sequence complementary to the target DNA, and the trans-activating crRNA (tracrRNA), which serves as a scaffold for Cas9 binding [2]. In practice, these are often combined into a single-guide RNA (sgRNA) for experimental simplicity [1].
A critical recognition element for the system is the Protospacer Adjacent Motif (PAM), a short (2-6 base pair) sequence adjacent to the target site that is essential for Cas9 recognition and binding [2]. For the most commonly used Cas9 from Streptococcus pyogenes (SpCas9), the PAM sequence is 5'-NGG-3' (where "N" is any nucleotide) [2]. The PAM requirement represents the primary constraint on target site selection for CRISPR-Cas9 experiments.
DNA Recognition and Cleavage Mechanism
The CRISPR-Cas9 genome editing process follows a sequential mechanism [1]:
Target Recognition: The guide RNA directs Cas9 to the target genomic locus through Watson-Crick base pairing between the guide sequence and the complementary DNA strand.
PAM Verification: Cas9 scans DNA for appropriate PAM sequences, which triggers local DNA melting and enables guide RNA-target DNA hybridization.
Conformational Activation: Successful target matching induces a conformational change in Cas9, activating its nuclease domains.
DNA Cleavage: The Cas9 protein contains two distinct nuclease domains: the HNH domain cleaves the DNA strand complementary to the guide RNA, while the RuvC-like domain cleaves the non-complementary strand, resulting in a precise double-strand break (DSB) [2].
The following diagram illustrates this molecular mechanism:
Cellular DNA Repair Pathways
Once Cas9 creates a double-strand break, the cell activates one of two major DNA repair pathways that determine the editing outcome [3] [1]:
Non-Homologous End Joining (NHEJ): This error-prone repair pathway directly ligates the broken DNA ends, often resulting in small insertions or deletions (indels) at the cleavage site. When these indels occur within a protein-coding exon, they can cause frameshift mutations that prematurely truncate the protein, effectively creating a gene knockout [1].
Homology-Directed Repair (HDR): This precise repair pathway uses a homologous DNA template to faithfully repair the break. Researchers can exploit this pathway by providing an exogenous donor DNA template to introduce specific sequence changes, enabling precise gene editing or correction [3].
For knockout cell line generation, the NHEJ pathway is typically leveraged to disrupt gene function through the introduction of frameshift mutations, making it the most commonly utilized pathway for functional gene knockout studies [1].
Quantitative Aspects of CRISPR-Cas9 Editing
Key Performance Metrics in Knockout Generation
The efficiency of CRISPR-Cas9 mediated knockout generation varies significantly based on multiple experimental factors. The following table summarizes critical quantitative metrics researchers must consider when designing knockout experiments:
Table 1: Key Performance Metrics for CRISPR-Cas9 Knockout Generation
| Metric | Typical Range | Influencing Factors | Optimization Strategies |
|---|---|---|---|
| Editing Efficiency | 5-90% indel rate [5] | gRNA design, chromatin accessibility, epigenetic marks, delivery method | Use optimized gRNA design tools; select gRNAs with high predicted scores [4] |
| Off-target Effects | Varies by gRNA specificity [6] | gRNA specificity, chromatin state, Cas9 variant | Use high-fidelity Cas9 variants; employ careful gRNA design with off-target prediction [5] |
| HDR Efficiency | Typically <10-20% of edited alleles [3] | Cell cycle stage, donor template design, competition with NHEJ | Synchronize cells in S/G2 phase; optimize donor design; use NHEJ inhibitors [3] |
| Delivery Efficiency | Varies by method and cell type [7] | Cell type, delivery method (viral, electroporation, lipid nanoparticles) | Match delivery method to cell type; optimize delivery conditions [8] [7] |
Comparison of CRISPR-Cas9 with Alternative Technologies
CRISPR-Cas9 has largely superseded earlier genome editing technologies due to its superior efficiency and ease of use. The table below compares key characteristics across major genome editing platforms:
Table 2: Comparison of Major Genome Editing Technologies
| Characteristic | CRISPR-Cas9 | TALENs | ZFNs |
|---|---|---|---|
| Targeting Molecule | RNA (gRNA) [1] | Protein (TALE domains) [1] | Protein (Zinc fingers) [1] |
| Target Recognition | 20-nt gRNA sequence [4] | 30-40 amino acids per base [1] | 3 bases per zinc finger [1] |
| Construction Time | Days [1] | Weeks [1] | Weeks [1] |
| Editing Efficiency | >80% in mammalian cells [1] | <30% typically [1] | <30% typically [1] |
| Multiplexing Capacity | High (multiple gRNAs) [1] | Limited | Limited |
| Technical Barrier | Low | High | High |
| Cost | Low | High | High |
Experimental Protocol for Knockout Cell Line Generation
The generation of knockout cell lines using CRISPR-Cas9 follows a systematic workflow that can be divided into four major phases. The following diagram provides a comprehensive overview of this process:
Detailed Stepwise Protocol
The following protocol provides detailed methodologies for generating knockout cell lines, with an estimated total timeline of 8-10 weeks [8]:
Target Selection: Identify the target exon within your gene of interest. For complete knockout, target constitutive exons present in all transcript variants. For isoform-specific knockout, target unique exons.
gRNA Design: Use bioinformatics tools (CHOPCHOP, Synthego CRISPR Design Tool, CRISPOR) to design gRNAs with:
Oligonucleotide Design: Design complementary oligonucleotides with appropriate overhangs for your CRISPR vector system (e.g., for LentiCRISPRv2: Forward: 5'-CACCG[N20]-3', Reverse: 5'-AAAC[N20]C-3') [7].
Annealing and Cloning:
- Phosphorylate and anneal oligonucleotides using T4 Polynucleotide Kinase
- Digest CRISPR vector with appropriate restriction enzyme (e.g., BbsI for LentiCRISPRv2)
- Ligate annealed oligos into linearized vector using T4 DNA Ligase
- Transform into competent cells (e.g., Stbl3 for lentiviral vectors) [7]
Plasmid Preparation: Isolve and purify high-quality plasmid DNA using endotoxin-free kits to ensure high transfection efficiency [8].
Determine Selection Conditions:
- Perform MTT assay to determine optimal antibiotic concentration (e.g., puromycin) that kills untransfected cells in 3-5 days
- Include untreated controls and serial dilutions of antibiotic [8]
Cell Transfection:
- For adherent cells: Seed cells to reach 70-80% confluence at transfection
- For suspension cells: Use lentiviral transduction (see specialized protocol below)
- Use lipid-based transfection (e.g., Lipofectamine 3000) or other appropriate method
- Incubate for 24-48 hours before starting selection [8]
Antibiotic Selection:
- Apply predetermined antibiotic concentration for 3-5 days
- Monitor cell death daily; replace selection media every 2-3 days
- Continue until all cells in negative control wells are dead [8]
Limiting Dilution:
- Trypsinize and count selected cell pool
- Dilute cells to concentration of 0.5 cells/100μL
- Plate 100μL per well in 96-well plates (theoretical probability: ≤1 cell/well)
- Include conditioned media (20-30%) to support single-cell growth [8]
Clone Expansion:
- Monitor wells weekly for single-cell derived colonies
- Expand positive wells gradually: 96-well → 24-well → 6-well → T25 flask
- Maintain detailed tracking of clone origins [8]
Genomic DNA Analysis:
- Extract genomic DNA from expanded clones
- PCR amplify target region
- Analyze by Sanger sequencing or next-generation sequencing
- Use trace decomposition software to detect indels [8]
Protein Validation:
- Perform western blot to confirm protein loss
- Use multiple antibodies targeting different protein domains when possible
- Include appropriate loading controls [8]
Functional Validation:
- Conduct functional assays specific to the target gene
- Examples: growth assays, differentiation capacity, drug sensitivity
- Compare to wild-type and heterozygous controls [8]
Specialized Protocol for Hard-to-Transfect Cells
For hard-to-transfect suspension cells (e.g., THP-1 immune cells), lentiviral delivery provides superior efficiency [7]:
Lentiviral Production:
- Co-transfect Lenti-X 293T cells with your CRISPR vector and packaging plasmids (psPAX2, pMD2.G)
- Collect viral supernatant at 48 and 72 hours post-transfection
- Concentrate virus using Lenti-X concentrator [7]
Viral Transduction:
- Determine viral titer using Lenti GoStix or other method
- Transduce target cells at appropriate MOI (typically 1-10) in the presence of polybrene (8μg/mL)
- Centrifuge plates (1000×g, 30-60 minutes) to enhance infection
- Begin antibiotic selection 48 hours post-transduction [7]
The Scientist's Toolkit: Essential Research Reagents
Successful generation of CRISPR-Cas9 knockout cell lines requires carefully selected reagents and tools. The following table outlines essential components for a typical knockout experiment:
Table 3: Essential Research Reagents for CRISPR-Cas9 Knockout Generation
| Reagent Category | Specific Examples | Function/Purpose | Considerations |
|---|---|---|---|
| CRISPR Vectors | pSpCas9(BB)-2A-Puro (PX459) [8], LentiCRISPRv2 [7] | Delivery of Cas9 and gRNA to cells; contains selection marker | Choose between transient (plasmid) and stable (lentiviral) expression |
| Restriction Enzymes & Cloning | BbsI (BpiI) [8], T4 DNA Ligase [8], T4 PNK [7] | gRNA insertion into CRISPR vector | BbsI creates compatible overhangs for gRNA oligo insertion |
| Delivery Reagents | Lipofectamine 3000 [8], Polybrene [7] | Facilitates cellular uptake of CRISPR components | Lipid-based for adherent cells; polybrene for viral transduction |
| Selection Agents | Puromycin [8] [7] | Selects for successfully transfected/transduced cells | Concentration must be optimized for each cell line |
| Cell Culture Reagents | Opti-MEM [8] [7], Fetal Bovine Serum [8] | Supports cell growth during and after editing | Reduced-serum media improves transfection efficiency |
| Validation Tools | Agarose [8], PVDF membranes [7], Antibodies for Western blot [7] | Confirms successful gene knockout at DNA and protein levels | Include wild-type controls for comparison |
| Bioinformatics Tools | CHOPCHOP [4], Synthego Design Tool [7], CRISPResso [4] | gRNA design and sequencing analysis | Assess both on-target efficiency and off-target potential |
Advanced Applications and Future Perspectives
AI-Driven CRISPR Design Advances
Recent advances in artificial intelligence have revolutionized CRISPR experimental design, addressing key challenges in efficiency and specificity [9] [5]. Machine learning models like DeepCRISPR and CRISPR-GPT can now predict guide RNA efficacy with high accuracy by analyzing sequence features, epigenetic markers, and chromatin accessibility data [5]. These tools significantly reduce the trial-and-error approach that has traditionally characterized CRISPR experiment optimization.
The development of OpenCRISPR-1, the first AI-generated CRISPR system, demonstrates the potential of this approach. This system, designed using large language models trained on over 1 million CRISPR operons, exhibits comparable or improved activity and specificity relative to SpCas9 while being 400 mutations away in sequence space [9]. Such AI-designed editors represent a new frontier in genome editing tools that bypass evolutionary constraints to generate editors with optimal properties [9].
Therapeutic Applications in Drug Development
CRISPR-Cas9 technology plays increasingly important roles in multiple stages of drug discovery and development [3] [1]:
Target Identification and Validation: CRISPR knockout screens enable genome-wide functional identification of genes essential for disease phenotypes, providing high-confidence therapeutic targets [3].
Disease Modeling: Isogenic cell lines with specific mutations can be rapidly generated to model disease states and test therapeutic interventions [3].
Cell Therapy Engineering: CRISPR-mediated knockout of immune checkpoint genes (e.g., PD-1) in CAR-T cells enhances their antitumor activity, demonstrating clinical potential in cancer immunotherapy [1].
The first FDA-approved CRISPR-based therapy, Casgevy, for sickle cell disease and β-thalassemia, validates the clinical potential of CRISPR technology and paves the way for future therapeutic applications in oncology and genetic disorders [1].
Addressing Technical Challenges
Despite its transformative potential, CRISPR-Cas9 application faces several technical challenges that active research continues to address:
Off-target Effects: Improvements in gRNA design algorithms, high-fidelity Cas9 variants, and novel detection methods have significantly reduced off-target editing [6] [5].
Delivery Efficiency: Advances in nanoparticle delivery systems, viral vectors, and physical methods (electroporation) continue to improve editing efficiency across diverse cell types [7] [2].
Editing Precision: Base editors and prime editors that enable precise single-base changes without double-strand breaks offer alternatives for applications requiring precision rather than gene disruption [2].
These ongoing developments ensure that CRISPR-Cas9 will remain at the forefront of genetic research and therapeutic development, with continued improvements in specificity, efficiency, and applicability across diverse biological contexts.
The CRISPR-Cas9 system has revolutionized biomedical research by providing an unparalleled tool for precise genome engineering. This technology enables researchers to create specific genetic modifications with unprecedented efficiency and has become indispensable across multiple domains, including functional genomics, disease modeling, and therapeutic development [10]. The core principle involves a programmable RNA-protein complex that introduces targeted double-strand breaks in DNA, facilitating gene knockout via non-homologous end joining (NHEJ) or precise gene correction through homology-directed repair (HDR) [10]. This protocol article details the application of CRISPR-Cas9 for generating knockout cell lines, with specific methodologies for target validation, disease modeling, and biologics development, providing researchers with optimized frameworks to advance their investigative and therapeutic goals.
Application Note 1: CRISPR-Cas9 for Target Validation
Core Principles and Workflow
Functional genomic screening using CRISPR-Cas9 knockout libraries enables the systematic identification and validation of novel therapeutic targets. Pooled CRISPR screens allow researchers to interrogate gene function at scale by assessing how individual gene knockouts affect cellular phenotypes, such as drug sensitivity, cell proliferation, or pathway activation [11]. The process involves transducing cells with a genome-wide or pathway-specific library of guide RNAs (gRNAs), selecting for desired phenotypes, and sequencing the gRNAs that become enriched or depleted to identify genes critical for the phenotype under investigation.
Table 1: CRISPR Knockout Efficiency Across Cell Types
| Cell Type | Editing Approach | Efficiency (INDEL%) | Key Optimization Parameters |
|---|---|---|---|
| Human Pluripotent Stem Cells (hPSCs) | Doxycycline-inducible Cas9 | 82-93% (single-gene); >80% (double-gene) | Cell-to-sgRNA ratio, nucleofection frequency, sgRNA stability [12] |
| THP-1 (Immune cells) | Lentiviral CRISPR-Cas9 | High (protocol-optimized) | Lentiviral transduction; validated sgRNA design [13] |
| General Mammalian Cell Lines | Plasmid or RNP transfection | Variable (3-4x more efficient than ZFN/TALEN) | Transfection method, sgRNA format, clonal selection [14] |
Detailed Protocol: Pooled CRISPR Screening for Target Identification
Materials and Reagents:
- Genome-wide or focused CRISPR knockout library (e.g., Brunello, GeCKO)
- Lentiviral packaging plasmids (psPAX2, pMD2.G)
- HEK293T cells for virus production
- Target cells of interest (ideally with stable Cas9 expression)
- Polybrene (8 μg/mL)
- Puromycin or appropriate selection antibiotic
- DNA extraction kit
- PCR purification kit
- Next-generation sequencing platform
Procedure:
Library Amplification and Virus Production:
- Transform the plasmid library into competent E. coli and culture on large-format LB agar plates with appropriate antibiotic to maintain library diversity.
- Isolate high-quality plasmid DNA using an endotoxin-free maxiprep kit.
- Co-transfect HEK293T cells with the library plasmid and packaging plasmids using PEI or commercial transfection reagents.
- Collect viral supernatant at 48 and 72 hours post-transfection, concentrate using PEG-it virus precipitation solution or ultracentrifugation, and titer using target cells.
Cell Transduction and Selection:
- Determine the multiplicity of infection (MOI) that results in approximately 30% transduction efficiency for your target cells to minimize multiple integrations.
- Transduce cells at a coverage of 500-1000x library representation (e.g., 500 million cells for a 100,000 gRNA library) with 8 μg/mL polybrene via spinfection (centrifugation at 1000 × g for 60-90 minutes at 32°C).
- Begin puromycin selection (concentration determined by kill curve) 48 hours post-transduction and maintain for 5-7 days until non-transduced control cells are completely dead.
Phenotypic Selection and Sequencing:
- Split cells into experimental and control groups (e.g., drug treatment vs. DMSO control) and maintain for 14-21 population doublings to allow phenotypic manifestation.
- Harvest at least 500 cells per gRNA for each condition to maintain library representation.
- Extract genomic DNA using a large-scale isolation method and amplify the integrated gRNA sequences with primers containing Illumina adapters and sample barcodes.
- Purify PCR products and quantify by qPCR before sequencing on an Illumina NextSeq or HiSeq platform.
Data Analysis:
- Align sequencing reads to the library reference and count gRNA reads for each sample.
- Use specialized algorithms (MAGeCK, CERES) to identify significantly enriched or depleted gRNAs between conditions, accounting for essential gene effects.
- Validate top hits using individual gRNAs in secondary functional assays.
CRISPR Screening Workflow for Target Identification
Application Note 2: Disease Modeling with CRISPR-Cas9
Advanced Disease Modeling Systems
CRISPR-Cas9 has dramatically advanced disease modeling by enabling precise introduction of pathogenic mutations into relevant cellular systems. Researchers can now create genetically accurate models that recapitulate disease pathophysiology across various platforms, including 2D cell cultures, 3D organoids, and organ-on-a-chip systems [15]. These models are particularly valuable for studying genetic disorders, cancer, and infectious diseases, allowing for investigation of disease mechanisms and high-throughput drug screening in human-relevant systems.
Table 2: CRISPR-Cas9 Disease Modeling Platforms
| Model System | Key Applications | CRISPR Editing Efficiency | Advantages |
|---|---|---|---|
| 2D Cell Cultures (primary cells, iPSCs) | High-throughput screening, pathway analysis | Variable: 20-93% depending on cell type and delivery method [12] | Simple, reproducible, amenable to HTS [15] |
| Organoids (iPSC-derived) | Developmental biology, genetic disorders, personalized medicine | Moderate to high in iPSCs with optimized protocols [12] [15] | 3D architecture, multicellular complexity, patient-specific [15] |
| Organ-on-a-Chip | Complex disease modeling, drug toxicity assessment | Requires pre-edited cells or specialized delivery | Physiological relevance, multi-organ interactions [15] |
| In Vivo Models | Preclinical validation, disease pathogenesis | Varies by delivery method and target tissue | Whole-organism context, systemic effects [15] |
Detailed Protocol: Generating Isogenic iPSC Lines for Disease Modeling
Materials and Reagents:
- Human induced pluripotent stem cells (iPSCs)
- Doxycycline-inducible Cas9 iPSC line (optional)
- Chemically modified synthetic sgRNAs (2'-O-methyl-3'-thiophosphonoacetate modifications)
- Nucleofector system (Lonza 4D-Nucleofector with P3 Primary Cell Kit)
- ROCK inhibitor (Y-27632)
- mTeSR or PGM1 medium
- Matrigel or Geltrex-coated plates
- Genomic DNA extraction kit
- T7E1 assay reagents or tracking of indels by decomposition (TIDE) analysis software
- Antibodies for pluripotency markers (OCT4, SOX2, NANOG)
Procedure:
sgRNA Design and Validation:
- Design 3-5 sgRNAs targeting your gene of interest using prediction tools (Benchling, CCTop) with high on-target and low off-target scores.
- Select sgRNAs targeting early exons to maximize probability of functional knockout.
- Validate sgRNA efficiency using a T7E1 assay or ICE analysis in a pilot experiment.
- Procure chemically modified synthetic sgRNAs with 2'-O-methyl-3'-thiophosphonoacetate modifications at both terminal to enhance stability [12].
Cell Preparation and Nucleofection:
- Culture iPSCs in PGM1 or mTeSR medium on Matrigel-coated plates until 80-90% confluent.
- Pre-treat cells with 10 μM ROCK inhibitor 1 hour before nucleofection.
- Dissociate cells with 0.5 mM EDTA to create single-cell suspension and count.
- Prepare nucleofection mixture: 8 × 10^5 cells, 5 μg sgRNA, and 100 μL P3 nucleofection buffer.
- Electroporate using Lonza 4D-Nucleofector with program CA-137 [12].
- Immediately transfer cells to pre-warmed medium with ROCK inhibitor in Matrigel-coated plates.
Recovery and Clonal Isolation:
- Change medium 24 hours post-nucleofection to remove ROCK inhibitor and dead cells.
- For iCas9 systems, add 2 μg/mL doxycycline for 48-72 hours to induce Cas9 expression.
- At 5-7 days post-nucleofection, harvest a portion of cells for initial editing efficiency assessment by T7E1 or TIDE.
- For clonal isolation, dissociate cells to single-cell suspension and seed at 0.5-1 cell/well in 96-well plates with conditioned medium and ROCK inhibitor.
- Expand clonal lines for 3-4 weeks with regular medium changes, monitoring colony morphology.
Genotypic and Phenotypic Validation:
- Extract genomic DNA from expanded clones and amplify target region by PCR.
- Confirm editing by Sanger sequencing and analyze using ICE or TIDE algorithms.
- For protein knockout confirmation, perform western blotting if suitable antibodies are available.
- Validate pluripotency retention by immunostaining for OCT4, SOX2, and NANOG.
- Bank at least 2-3 independent clones with confirmed knockout for downstream experiments.
iPSC Disease Model Generation Workflow
Application Note 3: CRISPR-Cas9 in Biologics Development
Therapeutic Applications and Clinical Progress
CRISPR-Cas9 has emerged as a transformative technology for biologics development, enabling creation of novel cell and gene therapies with demonstrated clinical efficacy. The first FDA-approved CRISPR-based therapy, CASGEVY, exemplifies this application—an ex vivo autologous cell therapy that edits the BCL11A gene in hematopoietic stem cells to treat sickle cell disease and transfusion-dependent beta thalassemia [16] [17]. Beyond ex vivo approaches, in vivo CRISPR therapies are advancing through clinical trials, utilizing lipid nanoparticle (LNP) delivery to target organs, particularly the liver, for conditions like hereditary transthyretin amyloidosis (hATTR) and hereditary angioedema (HAE) [16].
Detailed Protocol: Engineering Allogeneic CAR-T Cells for Oncology
Materials and Reagents:
- Healthy donor PBMCs or T-cell lines
- CRISPR-Cas9 ribonucleoprotein (RNP) complexes
- Synthetic sgRNAs targeting TRAC, B2M, and/or PDCD1
- CAR transgene construct (lentiviral or retroviral)
- Retronectin or Recombinant fibronectin fragment
- IL-2 and IL-7/IL-15 cytokines
- Anti-human CD3 and CD28 antibodies for activation
- Flow cytometry antibodies for CD3, CD19, CAR detection
- Cytotoxicity assay reagents (LDH, luciferase-based)
Procedure:
T-Cell Activation:
- Isolate PBMCs from leukapheresis product of healthy donor by Ficoll density gradient centrifugation.
- Activate T-cells using anti-CD3/CD28 antibodies (1 μg/mL each) in X-VIVO 15 or TexMACS medium with 5% human AB serum.
- Culture at 1-2 × 10^6 cells/mL in 24-well plates for 24-48 hours at 37°C, 5% CO2.
CRISPR-Cas9 RNP Electroporation:
- Design sgRNAs to disrupt endogenous T-cell receptor (TRAC), MHC-I (B2M), and/or checkpoint receptors (PD-1).
- Form RNP complexes by incubating 60 pmol Cas9 protein with 120 pmol synthetic sgRNA for 10-20 minutes at room temperature.
- Harvest activated T-cells, wash with PBS, and resuspend at 50-100 × 10^6 cells/mL in electroporation buffer.
- Mix 20 μL cell suspension with 5 μL RNP complex and electroporate using Lonza 4D-Nucleofector (program EO-115 for primary T-cells).
- Immediately transfer cells to pre-warmed complete medium with IL-2 (100 IU/mL).
CAR Transgene Delivery:
- 24 hours post-electroporation, transduce cells with CAR-encoding lentivirus at MOI 5-10 in retronectin-coated plates (centrifuge at 2000 × g for 90 minutes at 32°C).
- Add fresh medium with IL-2 (100 IU/mL) and IL-7/IL-15 (10 ng/mL each) after transduction.
- Expand cells for 10-14 days, maintaining density at 0.5-2 × 10^6 cells/mL with regular feeding.
Functional Validation:
- Assess editing efficiency by flow cytometry for CD3 (TRAC knockout) and MHC-I (B2M knockout) at day 5-7 post-electroporation.
- Evaluate CAR expression using protein L staining or target antigen staining.
- Perform functional assays including:
- Cytotoxicity against antigen-positive target cells (e.g., NALM-6 for CD19-CAR) using real-time cell analysis or luciferase-based killing assays.
- Cytokine secretion (IFN-γ, IL-2) upon antigen stimulation by ELISA or multiplex array.
- Exhaustion profiling after repeated antigen stimulation (PD-1, TIM-3, LAG-3 expression).
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for CRISPR-Cas9 Knockout Cell Line Generation
| Reagent Category | Specific Examples | Function & Application Notes |
|---|---|---|
| CRISPR Nucleases | spCas9, HiFi Cas9, Cas12a | DNA cleavage; HiFi Cas9 reduces off-target effects [10] |
| Guide RNA Formats | Chemically modified synthetic sgRNA, IVT sgRNA, lentiviral sgRNA | Targets Cas9 to specific genomic loci; chemical modifications enhance stability [12] |
| Delivery Systems | Lentivirus, electroporation (RNP), lipid nanoparticles (LNPs) | Introduces editing components into cells; choice depends on cell type [13] [16] |
| Validation Tools | T7E1 assay, TIDE analysis, ICE analysis, NGS | Confirms editing efficiency and characterizes mutations [12] [14] |
| Cell Culture Supplements | ROCK inhibitor (Y-27632), CloneR, RevitaCell | Enhances viability of sensitive cells (e.g., iPSCs, primary cells) after editing [12] |
| Selection Agents | Puromycin, blasticidin, fluorescence-activated cell sorting (FACS) | Enriches for successfully edited cells [13] [14] |
Technical Considerations and Troubleshooting
Optimizing Editing Efficiency
Achieving high editing efficiency requires careful optimization of multiple parameters. For difficult-to-transfect cells like THP-1 immune cells or primary cells, lentiviral delivery often provides superior results compared to electroporation or transfection [13]. In human pluripotent stem cells, systematic optimization of cell tolerance to nucleofection stress, sgRNA stability, nucleofection frequency, and cell-to-sgRNA ratio has enabled INDEL efficiencies of 82-93% for single-gene knockouts and over 80% for double-gene knockouts [12]. Chemical modifications to sgRNAs (2'-O-methyl-3'-phosphonoacetate) significantly enhance stability and editing efficiency [12].
Validation and Quality Control
Comprehensive validation of CRISPR knockout cell lines is essential for reliable experimental outcomes. DNA-level validation through Sanger sequencing coupled with ICE or TIDE analysis provides quantification of editing efficiency but should be complemented by protein-level validation through western blotting or flow cytometry when suitable antibodies are available [12] [14]. Researchers should be aware that high INDEL percentages at the DNA level do not always correlate with complete protein knockout, as reading frame shifts do not necessarily guarantee premature stop codons or degraded protein [12]. Functional assays specific to the target gene should ultimately confirm loss of function.
CRISPR-Cas9 technology has established itself as an indispensable tool across the biomedical research and therapeutic development spectrum. From systematic target validation through pooled screening approaches to creating genetically precise disease models and engineering novel biologic therapeutics, the applications detailed in this protocol article provide a framework for researchers to advance their investigative and therapeutic goals. As the field continues to evolve, with improvements in editing precision, delivery systems, and validation methodologies, CRISPR-based approaches will undoubtedly remain central to both fundamental biological discovery and the development of next-generation therapeutics.
Market Dynamics and Growth Drivers in the Gene Knockout Service Sector
Application Note: Market Landscape and Quantitative Analysis
The gene knockout cell line construction service market is experiencing significant growth, driven by the widespread adoption of CRISPR-Cas9 technology and its critical role in functional genomics and drug discovery. These services provide researchers with stable, genetically modified cell models essential for studying gene function, validating therapeutic targets, and understanding disease mechanisms.
Market Size and Growth Projections
The global market for gene knockout cell line construction is on a substantial growth trajectory, with complementary data from the broader gene editing market providing context for this expansion.
Table 1: Gene Knockout and Gene Editing Market Size Projections
| Market Segment | Base Year Value | Projected Year Value | Compound Annual Growth Rate (CAGR) | Time Period |
|---|---|---|---|---|
| Gene Knockout Cell Line Construction Service Market [18] | USD 571 Million (2024) | USD 799 Million (2031) | 5.1% | 2024-2031 |
| Gene Knockout Cell Line Construction Service Market [19] | - | - | 6.2% | 2025-2032 |
| Overall Gene Editing Market [20] | USD 11.29 Billion (2025) | USD 42.13 Billion (2034) | 15.76% | 2025-2034 |
Market Share and Segment Analysis
The market structure reveals distinct preferences in technology, service types, and end-users, with CRISPR-Cas9 dominating due to its precision, efficiency, and versatility [21] [19].
Table 2: Gene Knockout Service Market Share by Key Segments (2024)
| Segment Category | Leading Sub-segment | Approximate Market Share | Fastest-Growing Sub-segment |
|---|---|---|---|
| Technology/Editing Approach | CRISPR/Cas9 | 60% [21] | CRISPR Variants [21] |
| Service Type | Standard Knockout Cell Line Services | 45% [21] | Knockout in Difficult/Primary/iPSC-Derived Cells [21] |
| Cell Type | Cancer/Immortalized Cell Lines | Dominant Share [21] | iPSC-Derived Cell Lines [21] |
| Deliverable/Format | Clonal KO Cell Lines | Leading Share [21] | CRISPR KO Libraries [21] |
| End User | Pharmaceutical & Biotechnology Companies | Nearly 50% [21] | Contract Research Organizations (CROs) & Screening Labs [21] |
| Geographic Region | North America | 48% [21] | Asia-Pacific [21] [20] |
Key Market Drivers and Opportunities
- Advancements in Disease Modeling and Therapies: The rising global prevalence of severe genetic and chronic disorders fuels the demand for highly efficacious biologics and reliable genetic disease models. Gene knockout cell lines are indispensable for studying disease pathways and validating potential drug targets efficiently [21].
- Strategic Collaborations and Funding: The market is strengthened by partnerships and significant investments. For instance, in 2025, Asimov partnered with Ottimo Pharma to develop cell lines for cancer therapeutics, and Arbor Biotechnologies secured $73.9 million in financing to advance its gene editing pipeline [21].
- Integration of Artificial Intelligence: AI algorithms are enhancing the precision, effectiveness, and speed of gene knockout workflows by enabling predictive modeling for vector design and CRISPR guide RNA selection. AI is also being coupled with newer editing modalities like base editing and prime editing to refine knockout strategies [21].
- Expansion into Complex Models: A significant opportunity lies in meeting the escalating demand for stable knockout cell lines derived from difficult-to-engineer primary cells and induced pluripotent stem cells (iPSCs). These models are increasingly vital for advanced disease modeling, personalized medicine, and cell-based immunotherapies [21].
Protocol: CRISPR-Cas9 Mediated Knockout Cell Line Generation
This protocol provides a detailed methodology for generating single-gene knockout cell lines using the CRISPR-Cas9 system, incorporating best practices for optimizing efficiency and validation. The procedures are adapted from established protocols for both standard and hard-to-transfect cell lines [13] [8].
Protocol Workflow
The diagram below outlines the complete experimental workflow for generating knockout cell lines, from guide RNA design to final validation.
Part 1: sgRNA Design and Vector Construction (Steps 1-23)
Objective: To design and clone highly specific single-guide RNAs (sgRNAs) into a CRISPR-Cas9 expression vector.
Materials:
- pSpCas9(BB)-2A-Puro (PX459) V2.0 vector or similar [8]
- Oligonucleotides for sgRNA template
- Restriction enzyme (e.g., BpiI/FD1014)
- T4 DNA Ligase
- DH5α competent E. coli
Procedure:
- sgRNA Design: Use robust bioinformatics tools (e.g., from [22]) to design sgRNAs with high on-target efficiency scores (e.g., Vienna Bioactivity CRISPR (VBC) scores) and minimal predicted off-target effects. Adhere to design criteria such as GC content and positioning within the target exon [8] [22].
- Oligo Annealing: Phosphorylate and anneal the synthesized forward and reverse oligonucleotides to form a double-stranded DNA insert [8].
- Vector Digestion: Digest the PX459 vector with the appropriate restriction enzyme (e.g., BpiI) to create compatible ends. Treat the linearized vector with alkaline phosphatase to prevent re-circularization [8].
- Ligation: Ligate the annealed oligo duplex into the prepared vector using T4 DNA Ligase.
- Transformation and Plasmid Preparation: Transform the ligation product into DH5α competent E. coli. Plate on ampicillin-containing LB agar. Select colonies, inoculate cultures, and extract high-quality, endotoxin-free plasmid DNA using a commercial kit [8].
Part 2: Cell Line Transfection and Selection (Steps 24-57)
Objective: To deliver the recombinant CRISPR plasmid into target cells and select successfully transfected cells.
Materials:
- Target cell line (e.g., HCT-116, THP-1)
- Lipofectamine 3000 or similar transfection reagent [8]
- Lentiviral packaging plasmids (for hard-to-transfect cells) [13]
- Puromycin
- Cell culture media and reagents
Procedure:
- Determine Selection Pressure: Perform an MTT assay or similar viability assay to determine the optimal killing concentration of puromycin (or other appropriate selection antibiotic) for your specific cell line. This ensures complete death of untransfected cells without excessively harming transfected ones [8].
- Cell Transfection/Transduction:
- For Standard Cell Lines (e.g., HCT-116): Seed cells to reach 70-80% confluency at transfection. Transiently transfect with the recombinant plasmid using a lipid-based method like Lipofectamine 3000 according to the manufacturer's instructions [8].
- For Hard-to-Transfect Cells (e.g., THP-1): Use lentiviral delivery for higher efficiency. Package the sgRNA/Cas9 construct into lentiviral particles in HEK293T cells. Concentrate the virus and transduce the target THP-1 cells in the presence of a transduction enhancer like polybrene [13].
- Antibiotic Selection: Begin antibiotic selection (e.g., with puromycin) 24-48 hours post-transfection/transduction. Maintain selection for 3-5 days, or until all cells in the negative control well have died [13] [8].
Part 3: Monoclonal Cell Isolation and Validation (Steps 58-74)
Objective: To isolate single-cell clones and validate the gene knockout at the genetic and protein levels.
Materials:
- 96-well plates
- Lysis buffer for genomic DNA extraction
- PCR reagents
- Sequencing primers
- Western blotting reagents
Procedure:
- Monoclonal Isolation via Limiting Dilution:
- Harvest the selected pool of cells.
- Serially dilute the cell suspension and seed into 96-well plates at a statistical density of approximately 0.5-1 cell per well in a conditioned medium.
- Monitor wells daily under a microscope to identify and mark wells containing exactly one single cell. Wells with zero or multiple cells should be excluded from the analysis to ensure clonality [8].
- Clonal Expansion: Allow single cells to proliferate for 2-3 weeks, expanding them sequentially from 96-well plates to 24-well plates, then to T25 flasks.
- Genotypic Validation:
- Screening: Extract genomic DNA from a portion of the expanded clonal population.
- PCR and Sequencing: Amplify the targeted genomic region by PCR and subject the product to Sanger sequencing. Analyze the sequencing traces for indels (insertions or deletions) causing frameshifts at the target site, comparing them to the wild-type sequence [8].
- Phenotypic Validation:
- Western Blotting: Confirm the absence of the target protein using Western blot analysis. This is a crucial functional validation of a successful knockout [13].
- Functional Assays: Perform additional assays relevant to the gene's function (e.g., growth assays, reporter assays) to confirm the loss-of-function phenotype.
Table 3: Key Research Reagent Solutions for CRISPR Knockout Generation
| Item | Function/Application | Example Products/Sources |
|---|---|---|
| CRISPR Vector | All-in-one plasmid expressing Cas9, sgRNA, and a selection marker. | pSpCas9(BB)-2A-Puro (PX459) from Addgene [8] |
| sgRNA Design Tool | Bioinformatics platform for designing specific sgRNAs with minimal off-target effects. | Tools benchmarked in [22] (e.g., VBC scoring) |
| Delivery Reagent | Facilitates the introduction of CRISPR constructs into cells. | Lipofectamine 3000 (for transfection) [8] |
| Lentiviral System | Enables high-efficiency gene delivery, especially in hard-to-transfect cell lines. | Lentiviral packaging plasmids and protocols [13] |
| Selection Antibiotic | Kills untransfected/non-transduced cells, enriching for edited cells. | Puromycin [13] [8] |
| Validated Control sgRNAs | Positive (targeting essential genes) and negative (non-targeting) controls for assay validation. | Available from commercial vendors (e.g., Synthego) [23] |
The gene knockout service sector is a dynamic and critical component of modern biomedical research and drug development. The continuous refinement of protocols, coupled with strategic market movements and technological innovations, ensures that these services will remain at the forefront of enabling discoveries in functional genomics and therapeutic development.
The advent of programmable gene-editing technologies has revolutionized molecular biology and therapeutic development, with CRISPR-Cas9 emerging as the most transformative platform among zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR-Cas9 systems. These technologies enable precise modifications to genomic DNA through targeted double-strand breaks (DSBs), which are subsequently repaired by endogenous cellular mechanisms such as non-homologous end joining (NHEJ) or homology-directed repair (HDR) [24] [25]. While all three systems achieve targeted genome editing, they differ significantly in their molecular mechanisms, ease of design, specificity, and practical implementation. CRISPR-Cas9 has gained predominant adoption in research and therapeutic contexts due to its unparalleled simplicity, efficiency, and versatility compared to earlier protein-based platforms [26] [27]. This application note provides a comparative analysis of these technologies within the specific context of CRISPR-Cas9 knockout cell line generation, detailing experimental protocols and practical considerations for research and drug development applications.
Comparative Technology Analysis
Molecular Mechanisms and Design
The fundamental distinction between these gene-editing platforms lies in their DNA recognition and cleavage mechanisms. ZFNs utilize engineered zinc finger proteins, where each domain recognizes a 3-base pair DNA sequence, fused to the FokI nuclease domain. Effective cleavage requires two ZFN monomers binding to opposite DNA strands with proper orientation and spacing to facilitate FokI dimerization [28] [25]. Similarly, TALENs employ transcription activator-like effector (TALE) proteins from plant pathogens, where each TALE repeat recognizes a single nucleotide, fused to the FokI nuclease. Like ZFNs, TALENs function as pairs requiring dimerization for DNA cleavage [28] [29].
In contrast, the CRISPR-Cas9 system utilizes a guide RNA (gRNA) molecule with ~20 nucleotide complementarity to the target DNA sequence, which directs the Cas9 nuclease to the genomic locus. Cas9 cleavage requires both successful gRNA:DNA hybridization and the presence of a protospacer adjacent motif (PAM, typically 5'-NGG-3' for standard Streptococcus pyogenes Cas9) adjacent to the target site [27] [28]. This RNA-mediated targeting mechanism eliminates the need for complex protein engineering, representing a paradigm shift in gene-editing accessibility.
Table 1: Fundamental Characteristics of Gene-Editing Technologies
| Feature | ZFNs | TALENs | CRISPR-Cas9 |
|---|---|---|---|
| DNA Recognition Mechanism | Protein-DNA (Zinc finger domains) | Protein-DNA (TALE repeats) | RNA-DNA (gRNA complementarity) |
| DNA Recognition Specificity | 3 base pairs per zinc finger domain | 1 base pair per TALE repeat | ~20 nucleotides via gRNA |
| Nuclease Component | FokI endonuclease | FokI endonuclease | Cas9 endonuclease |
| Dimerization Requirement | Required (obligate heterodimer) | Required (obligate heterodimer) | Not required (single nuclease) |
| PAM Requirement | None | None | Required (varies by Cas9 variant) |
| Target Design Complexity | High (context-dependent effects) | Moderate (modular but repetitive) | Low (simple base pairing rules) |
Performance and Practical Considerations
CRISPR-Cas9 demonstrates significant advantages in design simplicity, efficiency, and cost-effectiveness. While ZFNs and TALENs require complex protein engineering that can take weeks to months with specialized expertise, CRISPR-Cas9 targets can be designed within days by simply modifying the 20-nucleotide gRNA sequence [26] [30]. The platform's efficiency in generating DSBs enables highly effective gene knockout through NHEJ-mediated indels, with modern systems achieving targeting efficiencies exceeding 70% in many cell types [27].
A critical advantage for knockout cell line generation is CRISPR-Cas9's capacity for multiplexed editing, allowing simultaneous disruption of multiple genes in a single experiment by introducing multiple gRNAs [30]. This capability is particularly valuable for modeling polygenic diseases or studying genetic interactions. Additionally, CRISPR-Cas9's cost profile is substantially lower than earlier technologies, making large-scale genetic screening approaches feasible [27].
Despite these advantages, CRISPR-Cas9 exhibits a higher propensity for off-target effects compared to TALENs, though advanced Cas9 variants with enhanced fidelity (e.g., HiFi Cas9, eCas9) have substantially mitigated this concern [27] [28]. TALENs maintain application niches requiring exceptional specificity, particularly for targets with challenging sequence contexts or minimal tolerance for off-target activity [26] [29].
Table 2: Performance Metrics for Gene-Editing Technologies in Knockout Cell Line Generation
| Parameter | ZFNs | TALENs | CRISPR-Cas9 |
|---|---|---|---|
| Development Timeline | 1-6 months [24] | ~1 month [24] | <1 week [24] |
| Relative Cost | High [27] | Medium [24] | Low [27] |
| Editing Efficiency | Moderate [26] | Moderate to High [26] | High [27] |
| Multiplexing Capacity | Limited [27] | Limited [27] | High (multiple gRNAs) [30] |
| Off-Target Effects | Lower than CRISPR [24] | Lower than CRISPR [24] | Moderate (improved with variants) [27] |
| Delivery Considerations | Compatible with viral vectors [27] | Challenging due to large size [24] | Highly compatible with multiple delivery methods [27] |
| Therapeutic Validation | Clinical success (e.g., HIV) [27] | Preclinical and niche applications [27] | Multiple clinical trials (e.g., β-thalassemia) [27] |
CRISPR-Cas9 Knockout Cell Line Development Workflow
CRISPR-Cas9 Knockout Cell Line Generation Protocol
Experimental Design and gRNA Selection
Effective CRISPR-Cas9 knockout begins with strategic gRNA design targeting early exons of the gene of interest to maximize probability of frameshift mutations. Target sequences should follow the pattern 5'-G(N)~19NGG-3' for SpCas9, where the 20-nucleotide guide sequence precedes the 3' NGG PAM [28]. Utilize bioinformatic tools to select gRNAs with minimal off-target potential by assessing genome-wide complementarity. Design multiple gRNAs (typically 3-4) targeting different regions to enhance knockout efficiency. Include bioinformatic analysis for potential off-target sites with up to 3-4 nucleotide mismatches, particularly in coding regions [27] [28].
For the homologous repair template (if applicable), design single-stranded oligodeoxynucleotides (ssODNs) with at least 40-base homology arms flanking the target site, incorporating desired mutations and silent restriction sites to facilitate screening. For knockout generation, error-prone NHEJ repair will introduce indels; no donor template is required [25].
Delivery Methods and Transfection
CRISPR-Cas9 components can be delivered as plasmid DNA, in vitro transcribed mRNA, or ribonucleoprotein (RNP) complexes. RNP delivery offers rapid action and reduced off-target effects by minimizing nuclease exposure [28]. For mammalian cells, utilize appropriate delivery methods:
- Lipofection: Complex CRISPR plasmids or RNPs with lipid-based transfection reagents following manufacturer protocols. Optimal for adherent cell lines (HEK293, HeLa).
- Electroporation: Apply electrical pulses to facilitate component entry. Superior for hard-to-transfect cells (primary cells, immune cells).
- Viral Delivery: Package gRNA and Cas9 into lentiviral or adenoviral vectors for sustained expression. Requires additional biosafety precautions [27].
Transfert cells at 70-80% confluence, including appropriate controls (non-targeting gRNA, mock transfection). For plasmid-based delivery, use a 1:1-1:3 mass ratio of Cas9:gRNA expression vectors [28].
Screening and Validation
After 48-72 hours post-transfection, assess editing efficiency via T7 Endonuclease I or Surveyor assay to detect mismatched heteroduplex DNA. Expand transfected cells under appropriate selection (e.g., puromycin for integrated selection markers) for 7-14 days to derive clonal populations [30].
Isolate single cells by fluorescence-activated cell sorting (FACS) or limiting dilution into 96-well plates. Expand clones over 2-3 weeks, then extract genomic DNA for PCR amplification of the target region. Analyze amplicons by sequencing to identify frameshift mutations. Confirm knockout at protein level via Western blotting or immunostaining [27].
Table 3: Essential Research Reagents for CRISPR-Cas9 Knockout Generation
| Reagent Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Cas9 Expression Systems | SpCas9 expression plasmids, mRNA, recombinant protein | Catalytic component for DNA cleavage; protein delivery reduces off-target effects [28] |
| gRNA Expression Vectors | U6-promoter driven gRNA cloning vectors | Guide RNA expression; modified vectors enable multiplexed targeting [28] |
| Delivery Reagents | Lipofectamine CRISPRMAX, electroporation systems | Facilitate cellular entry of editing components; choice depends on cell type [27] |
| Validation Enzymes | T7 Endonuclease I, Surveyor nuclease | Detect indel mutations in pooled populations prior to clonal isolation [30] |
| Selection Agents | Puromycin, G418, fluorescent markers | Enrich for successfully transfected cells; critical for low-efficiency deliveries [27] |
| Clonal Isolation Tools | 96-well plates, FACS systems, limiting dilution equipment | Establish monoclonal cell populations from edited pools [30] |
CRISPR-Cas9 Gene Editing Mechanism
Applications in Drug Discovery and Development
CRISPR-Cas9 knockout cell lines have become indispensable tools in pharmaceutical research, enabling systematic functional genomic screening to identify novel drug targets and elucidate mechanisms of action [27]. Pooled CRISPR libraries allow genome-wide loss-of-function screens that identify genetic dependencies and synthetic lethal interactions for therapeutic exploitation. In target validation, isogenic knockout cell lines provide definitive evidence for target necessity in disease-relevant phenotypes [27].
The technology has accelerated the development of biologically relevant disease models, including patient-derived organoids with engineered mutations that recapitulate human disease pathophysiology. These advanced models improve preclinical prediction of drug efficacy and toxicity [27]. Additionally, CRISPR-Cas9 enables investigation of drug resistance mechanisms through deliberate introduction of resistance-associated mutations, facilitating the design of next-generation therapeutics that overcome common resistance pathways [27].
CRISPR-Cas9's clinical translation has already demonstrated remarkable success in ex vivo gene therapies for monogenic disorders, with approved treatments for sickle cell anemia and β-thalassemia representing paradigm-shifting applications of the technology [27]. The platform's versatility continues to expand through engineered Cas variants with altered PAM specificities, reduced off-target activity, and novel functionalities including base editing and epigenetic modulation [31].
CRISPR-Cas9 represents a superior gene-editing platform for knockout cell line generation compared to ZFNs and TALENs, offering unprecedented design simplicity, efficiency, and multiplexing capability. While older technologies maintain relevance for specific applications requiring exceptional precision or challenging targeting contexts, CRISPR-Cas9's versatility and accessibility have democratized genetic engineering across biological research and therapeutic development. The continued evolution of CRISPR-based technologies, including enhanced specificity systems and novel editing modalities, promises to further accelerate drug discovery and expand therapeutic possibilities. Researchers generating knockout cell lines for mechanistic studies or drug development should prioritize CRISPR-Cas9 as the default technology while implementing appropriate controls and validation strategies to ensure experimental rigor.
Optimized CRISPR Workflow: A Step-by-Step Protocol from sgRNA Design to Clonal Expansion
Strategies for High-Efficiency sgRNA Design and the Role of AI-Powered Tools
The generation of CRISPR-Cas9 knockout cell lines represents a cornerstone technique in modern genetic research and drug development. The efficiency of this process hinges critically on the design of the single-guide RNA (sgRNA), which directs the Cas9 nuclease to its specific genomic target. An optimal sgRNA must fulfill two essential requirements: high on-target efficiency to ensure effective gene knockout and minimal off-target effects to prevent unintended genomic alterations that could compromise experimental results or therapeutic safety [32] [33].
The evolution of sgRNA design has progressed from early empirical rules to sophisticated artificial intelligence (AI)-driven models capable of predicting sgRNA activity with remarkable accuracy. These advancements are particularly crucial within the context of knockout cell line generation for drug discovery, where reproducibility, precision, and time efficiency are paramount. This application note delineates structured strategies for high-efficiency sgRNA design and examines the transformative role of AI-powered tools in optimizing CRISPR-Cas9 workflows for research and therapeutic development.
Core Principles of High-Efficiency sgRNA Design
Sequence-Specific Determinants of sgRNA Activity
The guide sequence itself is a primary determinant of sgRNA efficacy. Research has identified specific sequence motifs that significantly impair CRISPR-Cas9 activity. Studies reveal that TT-motifs and GCC-motifs located at the 3' end of the targeting sequence (the PAM-proximal region) are strongly associated with reduced knockout efficiency [34]. The presence of these motifs can diminish editing efficiency by up to ten-fold, establishing them as critical avoidance criteria in sgRNA selection [34].
Beyond specific inhibitory motifs, general sequence characteristics contribute to sgRNA performance. While optimal GC content typically falls between 40-60%, this parameter alone is insufficient for predicting efficacy and must be considered alongside other sequence features [33]. The positioning of the sgRNA target site within the gene structure also influences functional outcomes; for protein-coding genes, targeting early exons increases the likelihood of generating frameshift mutations that result in complete gene knockout [33].
Structural Optimization of sgRNA
The non-guide portion of the sgRNA molecule, known as the scaffold, significantly influences transcriptional efficiency and stability. Systematic investigations demonstrate that modifying the sgRNA scaffold by extending the duplex length by approximately 5 base pairs and mutating the fourth thymine (T) in the consecutive T-stretch to cytosine (C) or guanine (G) markedly enhances knockout efficiency across diverse cell types [35]. This optimized structure alleviates limitations imposed by RNA polymerase III pausing at poly-T sequences while potentially improving sgRNA stability or Cas9 binding affinity.
Table 1: Key Parameters for High-Efficiency sgRNA Design
| Parameter | Optimal Characteristic | Biological Rationale | Experimental Validation |
|---|---|---|---|
| Inhibitory Motifs | Avoid TT- and GCC-motifs in seed region | These motifs interfere with Cas9 binding or cleavage activity [34] | 10-fold reduction in knockout efficiency observed with motif presence [34] |
| GC Content | 40-60% | Balanced stability; avoids overly stable/unstable binding [33] | Empirical observation from high-throughput screens [33] |
| scaffold Structure | Extended duplex (+5 bp) + T→C/G mutation at position 4 | Prevents RNA pol III pausing; improves complex stability [35] | Significant efficiency improvement across 16 sgRNAs in multiple cell lines [35] |
| Target Location | Early coding exons | Maximizes probability of frameshift/nonsense mutations | Standard practice for knockout generation |
| PAM Specificity | NGG for SpCas9 | Essential for Cas9 recognition and binding [33] | Fundamental to CRISPR-Cas9 system specificity |
AI-Powered Tools for Predictive sgRNA Design
Evolution of Predictive Algorithms
The development of AI-driven sgRNA design tools represents a paradigm shift from reliance on simple sequence rules to data-intensive predictive modeling. Early rule-based systems have evolved into sophisticated machine learning (ML) and deep learning (DL) frameworks trained on large-scale experimental datasets encompassing thousands of sgRNA activity measurements [36] [33] [37].
Significant algorithmic milestones include:
- Rule Set 1/2/3: Progressive iterations developed by Doench et al., with each version incorporating larger training datasets and more sophisticated modeling approaches, culminating in Rule Set 3 which accounts for tracrRNA sequence variations [36] [33].
- CRISPRscan: A model trained on in vivo efficiency data from zebrafish embryos, highlighting the importance of species-specific and context-dependent factors [36] [33].
- DeepSpCas9: A convolutional neural network (CNN) model demonstrating superior generalization across diverse datasets compared to previous algorithms [36].
- CRISPRon: A DL framework that integrates both sgRNA sequence features and epigenomic information such as chromatin accessibility to predict Cas9 editing efficiency with enhanced accuracy [38].
Integration of AI Tools in Experimental Workflows
AI-powered sgRNA design platforms have become accessible through user-friendly web interfaces that provide comprehensive efficiency and specificity scoring. These tools employ diverse algorithmic approaches to balance on-target and off-target predictions:
Table 2: Comparison of Major AI-Powered sgRNA Design Tools
| Tool | Key Algorithms | Strengths | Applications in Knockout Cell Line Generation |
|---|---|---|---|
| CRISPick | Rule Set 3 (on-target), CFD (off-target) [33] | Regularly updated algorithms; user-friendly interface | Primary sgRNA selection with specificity scoring |
| CHOPCHOP | Multiple scoring systems (Rule Set, CRISPRscan) [33] | Versatility for different CRISPR systems; visual output | Target site visualization and multi-system design |
| CRISPOR | Combines multiple on-target and off-target scores [33] | Detailed off-target analysis with mismatch tolerance | Comprehensive specificity profiling for critical applications |
| GenScript Tool | Rule Set 3, CFD [33] | Integrated with ordering system; overall score balancing | Rapid design-to-execution pipeline |
These platforms typically generate multiple candidate sgRNAs for each target gene, ranking them based on composite scores that integrate predicted on-target efficiency, off-target potential, and other relevant features. Researchers can then select the top-performing guides for experimental validation, significantly increasing the success rate of knockout cell line generation.
Diagram 1: AI-Enhanced sgRNA Design Workflow. This workflow integrates computational design with AI-powered analysis for selecting high-efficiency sgRNAs with minimal off-target risk.
Advanced AI Implementations: CRISPR-GPT and Agentic Automation
The integration of large language models (LLMs) with CRISPR expertise represents the frontier of AI-powered genome engineering. CRISPR-GPT, developed at Stanford Medicine, is a specialized AI system that functions as an experimental co-pilot, assisting researchers in designing, optimizing, and troubleshooting CRISPR experiments through natural language interactions [39] [40].
This agentic AI system employs a multi-agent architecture with specialized components:
- Planner Agent: Deconstructs user experimental goals into logical workflows
- Task Executor Agent: Automates specific design steps using state machines
- User-Proxy Agent: Facilitates natural language communication with researchers
- Tool Provider Agents: Integrate peer-reviewed literature and bioinformatic resources [40]
In practical validation, researchers using CRISPR-GPT achieved ~80% editing efficiency in knocking out four genes (TGFβR1, SNAI1, BAX, BCL2L1) in A549 lung cancer cells on their first attempt, dramatically reducing the traditional trial-and-error周期 [39] [40]. Similarly, the system guided successful epigenetic activation in melanoma cells with up to 90% efficiency [40].
CRISPR-GPT incorporates critical safety features including dual-use risk mitigation, human germline editing restrictions, and privacy safeguards for identifiable genetic sequences, addressing essential ethical considerations in AI-driven genome editing [40].
Experimental Protocol for sgRNA Validation in Knockout Cell Line Generation
sgRNA Design and Computational Validation
Target Identification: Select target sequences within early exons of the gene of interest, ensuring presence of PAM sequence (NGG for SpCas9) immediately 3' to the 20nt target site [33].
AI-Guided Design:
- Input target genomic sequence into CRISPick or alternative design platform
- Select top 3-5 sgRNA candidates based on composite scores (prioritizing Rule Set 3 >0.6)
- Perform comprehensive off-target analysis using CFD scoring (threshold <0.05) [33]
sgRNA Construct Preparation:
- Synthesize sgRNA expression cassettes with optimized scaffold (T→C mutation at position 4 + 5 bp duplex extension) [35]
- Clone into appropriate delivery vector (lentiviral, plasmid)
Experimental Validation and Screening
Cell Transfection/Transduction: Deliver sgRNA-Cas9 constructs to target cells using optimized method (lipofection, electroporation, viral transduction).
Efficiency Assessment (72-96 hours post-delivery):
- T7 Endonuclease I Assay: Qualitative detection of indel mutations
- Next-Generation Sequencing: Quantitative analysis of editing efficiency and indel spectrum
- Flow Cytometry: For genes affecting surface markers [35]
Clonal Selection and Validation:
- Single-cell sorting or limiting dilution to establish clonal populations
- Genomic DNA extraction and PCR amplification of target locus
- Sanger sequencing with TIDE decomposition or NGS to verify bi-allelic knockout
- Western blot or immunostaining to confirm protein loss
Diagram 2: Experimental Workflow for Knockout Cell Line Generation. This protocol outlines key steps from computational design to experimental validation of CRISPR-Cas9 knockout cell lines.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Cas9 Knockout Cell Line Generation
| Reagent Category | Specific Examples | Function in Workflow | Optimization Notes |
|---|---|---|---|
| Cas9 Expression System | SpCas9 expression plasmid, Stable Cas9-expressing cell lines | Provides nuclease activity for DNA cleavage | Codon-optimized versions enhance efficiency in mammalian cells |
| sgRNA Expression Vectors | Lentiviral vectors (pLKO, pLentiGuide), All-in-one systems | Deliver sgRNA to target cells; enable stable expression | U6 promoter-driven transcription; modified scaffolds boost activity [35] |
| Delivery Reagents | Lipofectamine, Polyethylenimine (PEI), Electroporation systems | Introduce CRISPR components into cells | Method selection depends on cell type and efficiency requirements |
| Validation Enzymes | T7 Endonuclease I, Surveyor Nuclease | Detect indel formation at target sites | Quick but qualitative; requires optimization of digestion conditions |
| Selection Markers | Puromycin, Blasticidin, Fluorescent proteins (GFP) | Enumerate transfected cells or select stable integrants | Antibiotic concentration must be predetermined for each cell line |
| NGS Library Prep Kits | Illumina CRISPR amplicon sequencing kits | Quantitative analysis of editing efficiency and indel spectrum | Enables precise quantification of knockout efficiency and off-target assessment |
The generation of high-quality CRISPR-Cas9 knockout cell lines for drug development and functional genomics requires a methodical approach to sgRNA design that integrates established sequence principles with cutting-edge AI tools. Strategic avoidance of inhibitory motifs, implementation of scaffold optimization, and leveraging of AI-powered prediction platforms collectively enhance editing efficiency while mitigating off-target risks.
The emergence of specialized AI systems like CRISPR-GPT demonstrates the potential for democratizing CRISPR expertise and accelerating therapeutic development cycles from years to months [39]. As these technologies continue to evolve, researchers in both academic and industrial settings stand to benefit from increased experimental success rates, reduced development timelines, and enhanced reproducibility in knockout cell line generation—ultimately advancing the discovery and validation of novel therapeutic targets.
The generation of CRISPR-Cas9 knockout cell lines is a cornerstone of modern functional genomics and drug discovery research. The success of these experiments depends critically on the efficient delivery of CRISPR components—Cas9 nuclease and single-guide RNA (sgRNA)—into the nucleus of target cells. The choice of delivery method directly influences editing efficiency, cellular viability, off-target effects, and experimental timelines. This application note provides a detailed comparative analysis of three principal delivery platforms—lipofection, electroporation, and viral transduction—framed within the context of knockout cell line generation. We present structured quantitative data, detailed protocols for each method, and strategic guidance to enable researchers to select and optimize the appropriate delivery system for their specific experimental needs, from initial proof-of-concept studies to the creation of clonal cell lines for downstream applications.
Comparative Analysis of Delivery Methods
The table below summarizes the key characteristics of the three primary delivery methods, providing a foundation for selection.
Table 1: Key Characteristics of CRISPR-Cas9 Delivery Methods
| Feature | Lipofection | Electroporation | Viral Transduction |
|---|---|---|---|
| Principle | Lipid-based complexes fuse with cell membrane [41] | Electrical pulses create transient pores in membrane [41] | Viral particles infect cells for gene delivery [42] |
| Typical Cargo | DNA, mRNA, RNP [42] | DNA, mRNA, RNP [43] [41] | DNA (LV, AdV), RNA (LV) [42] |
| Editing Efficiency | Variable; highly cell-type dependent | High; e.g., >90% INDELs in HSPCs [43] | High; stable genomic integration [13] |
| Cellular Toxicity | Moderate | Higher, requires parameter optimization [44] [45] | Lower, but biosafety concerns exist [42] |
| Transgene Expression | Transient | Transient (for RNP/mRNA) | Stable, long-term [42] |
| Throughput | High | Medium to High | Medium |
| Technical Skill | Low | Medium | Medium to High |
| Cost | Low | Medium | High |
Detailed Methodologies and Protocols
Lipofection
Lipofection uses cationic lipids or lipid nanoparticles to form complexes with nucleic acids or proteins, which fuse with the cell membrane and release their cargo into the cytoplasm [42]. It is a cost-effective and high-throughput method suitable for a wide range of immortalized cell lines.
Protocol: Lipofection of CRISPR RNP Complexes using Lipofectamine CRISPRMAX [45]
- Step 1: RNP Complex Formation
- Complex 3 µg of purified Cas9 protein with synthetic sgRNA at a 1:2.5 mass ratio (Cas9:sgRNA).
- Incubate at room temperature for 10-20 minutes to form the RNP complex.
- Step 2: Lipid Complex Preparation
- Dilute the RNP complex in an appropriate serum-free medium.
- In a separate tube, dilute Lipofectamine CRISPRMAX reagent in serum-free medium.
- Combine the diluted RNP and diluted lipid reagent at a 1:1 ratio. Mix gently and incubate for 10-15 minutes at room temperature to form lipid nanoparticles.
- Step 3: Cell Transfection
- Aspirate the culture medium from cells (e.g., HEK293, HeLa) at 70-80% confluency and wash with PBS.
- Add the lipid nanoparticle complexes dropwise onto the cells.
- Incubate cells at 37°C, 5% CO₂ for 4-6 hours before replacing with fresh complete medium.
- Step 4: Analysis
- Assess editing efficiency 48-72 hours post-transfection via genomic DNA extraction, PCR, and T7E1 assay or sequencing.
Research Reagent Solutions for Lipofection
| Item | Function/Description |
|---|---|
| Lipofectamine CRISPRMAX | A proprietary lipid reagent specifically formulated for efficient RNP delivery [45]. |
| Opti-MEM | A serum-free medium used for diluting lipids and cargo, minimizing complex disruption. |
| Chemical Enhancers (e.g., Nedisertib) | DNA-PK inhibitors that can be added post-transfection to improve Homology-Directed Repair (HDR) efficiency by ~24% [46]. |
Electroporation
Electroporation utilizes electrical pulses to create transient pores in the cell membrane, allowing CRISPR cargo to enter the cell directly. Nucleofection is a specialized form of electroporation optimized for nuclear delivery, making it highly effective for hard-to-transfect cells like primary cells and stem cells [41].
Protocol: Nucleofection of RNP Complexes in Human Pluripotent Stem Cells (hPSCs) [47]
- Step 1: RNP Complex Formation
- Resuspend chemically synthesized, modified sgRNA (CSM-sgRNA) and Cas9 protein in nuclease-free water. A typical reaction uses 5 µg of sgRNA for 8x10⁵ cells [47].
- Step 2: Cell Preparation
- Harvest hPSCs using EDTA or a gentle cell dissociation reagent to create a single-cell suspension.
- Centrifuge and resuspend the cell pellet in the appropriate Nucleofector Solution, specific to the cell type (e.g., P3 Primary Cell 4D-Nucleofector X Kit).
- Step 3: Nucleofection
- Mix the cell suspension with the pre-formed RNP complex.
- Transfer the mixture to a nucleocuvette and electroporate using a device-specific program (e.g., program CA-137 for hPSCs on a Lonza 4D-Nucleofector) [47].
- Immediately after pulsing, add pre-warmed culture medium to the cuvette and transfer the cells to a culture plate pre-coated with Matrigel.
- Step 4: Post-Transfection Recovery
- Consider a repeated nucleofection 3 days after the first round to boost INDEL efficiency, which can reach 82-93% for single-gene knockouts [47].
- Validate knockout via colony PCR, Sanger sequencing, and Western blotting.
Diagram 1: Electroporation workflow for CRISPR-Cas9 delivery, highlighting key steps for high efficiency in sensitive cells.
Research Reagent Solutions for Electroporation
| Item | Function/Description |
|---|---|
| 4D-Nucleofector System (Lonza) | Instrument with pre-optimized programs for specific cell types (e.g., program DZ-100 for BEL-A cells) [46]. |
| Cell Type-Specific Kits | Solutions and reagents (e.g., P3 Primary Cell Kit) formulated for different cell lines to maximize viability and efficiency. |
| Chemically Modified sgRNA | sgRNA with 2’-O-methyl-3'-thiophosphonoacetate modifications at both ends to enhance stability within cells [47]. |
Viral Transduction
Viral transduction employs engineered viruses to deliver CRISPR cassettes. Lentiviral vectors (LVs) are particularly valuable for hard-to-transfect suspension cell lines (e.g., THP-1) and when stable, long-term expression is required, as they integrate into the host genome [13] [42].
Protocol: Lentiviral Knockout in THP-1 Cells [13]
- Step 1: sgRNA Cloning and Viral Production
- Design and clone sgRNA sequences into a lentiviral CRISPR vector (e.g., lentiCRISPRv2).
- Co-transfect the packaging plasmid, envelope plasmid (e.g., VSV-G), and the transfer vector into a producer cell line (e.g., HEK293T) using a method like lipofection.
- Collect the viral supernatant 48 and 72 hours post-transfection. Concentrate the virus if necessary by ultracentrifugation.
- Step 2: Cell Transduction
- Seed THP-1 cells in growth medium supplemented with polybrene (6-8 µg/mL) to enhance viral infection.
- Add the concentrated lentiviral supernatant to the cells and centrifuge (e.g., 2000 x g for 2 hours at 32°C) to spinfect the cells, significantly improving transduction efficiency.
- Incubate the cells for 24-48 hours.
- Step 3: Selection and Validation
- Replace the medium with fresh medium containing an appropriate antibiotic (e.g., puromycin) to select for successfully transduced cells.
- Maintain selection for at least 3-7 days until control cells are dead.
- Expand the polyclonal population and validate knockout via sequencing and Western blot analysis for the target protein (e.g., GSDMD) [13].
Research Reagent Solutions for Viral Transduction
| Item | Function/Description |
|---|---|
| VSV-G Pseudotyped Lentivirus | A common pseudotype that confers broad tropism and high stability, utilizing the LDL receptor for entry [48]. |
| Polybrene | A cationic polymer that reduces charge repulsion between viral particles and the cell membrane, increasing transduction efficiency. |
| LDLR Knockout Producer Cells | Engineered HEK293T cells with the LDL receptor knocked out to prevent "retro-transduction," which can improve viral yield by reducing producer cell infection [48]. |
Advanced Considerations and Troubleshooting
Cargo Selection and Its Impact
The format of the CRISPR components significantly influences the editing outcome and specificity.
Table 2: Impact of CRISPR Cargo Format on Editing Outcomes [43] [42]
| Cargo Format | Pros | Cons | Ideal Delivery Method |
|---|---|---|---|
| Plasmid DNA | Low cost, easy to manipulate [42]. | Risk of random integration, prolonged Cas9 expression increases off-targets, cytotoxicity [42]. | Lipofection, Viral Transduction. |
| Cas9 mRNA + gRNA | Rapid translation, transient expression, lower immunogenicity than DNA [43]. | Requires nuclear entry for efficiency, less stable than DNA [41]. | Lipofection, Electroporation. |
| Ribonucleoprotein (RNP) | Immediate activity, shortest exposure, highest specificity, reduced off-target effects [43] [42]. | More expensive, requires purified protein. | Electroporation (gold standard), Lipofection. |
Method Selection Workflow
Choosing the optimal delivery method requires a systematic approach based on cell type and experimental goals. The following decision tree provides a strategic framework for selection.
Diagram 2: A strategic decision workflow for selecting the optimal CRISPR delivery method based on cell type and experimental requirements.
Addressing Common Challenges
- Low Editing Efficiency in Lipofection: Ensure cargo-lipid complexes are formed at the correct ratio in serum-free medium. Pre-test different lipid reagents on your specific cell line. For precise edits, consider adding HDR enhancers like Nedisertib [46].
- Poor Cell Viability in Electroporation: This is often due to suboptimal electrical parameters. Use cell-type-specific protocols and kits. Ensure cells are healthy before nucleofection and that recovery medium is added immediately after the pulse. Titrating the RNP concentration can also help balance efficiency and viability [47] [45].
- Mosaicism in Viral Transduction: This can occur if Cas9 expression persists. Using RNP delivery via electroporation is the most effective way to avoid mosaicism by ensuring transient Cas9 activity. For viral methods, self-inactivating (SIN) vectors and inducible Cas9 systems can help limit the editing window.
The generation of clonal CRISPR-Cas9 knockout (KO) cell lines represents a cornerstone technique in modern molecular biology, enabling the precise dissection of gene function in health and disease. For researchers and drug development professionals, this process provides a critical tool for validating therapeutic targets, understanding disease mechanisms, and producing reliable, genetically defined cellular models. The complete workflow integrates three distinct but interdependent technical phases: the delivery of CRISPR components into cells via transfection, the isolation of single cells to ensure clonality, and the subsequent expansion of these cells into stable, genetically homogeneous populations [49] [14].
Each phase presents unique challenges that can compromise the entire experiment if not properly optimized. Transfection efficiency varies significantly between cell types and reagents, single-cell isolation is a technically demanding and low-yield process, and clonal expansion requires meticulous culture conditions to prevent stress-induced phenotypes or cell death [8] [14]. This application note provides a detailed, step-by-step protocol framed within the context of CRISPR-Cas9 knockout generation, incorporating quantitative data and structured workflows to guide researchers through this complex process.
Strategic Planning and Knockout Design
Before initiating laboratory work, careful strategic planning is essential for success. The first decision involves selecting a CRISPR delivery system. The table below compares the primary options.
Table 1: Comparison of CRISPR/Cas9 Delivery Systems for Knockout Generation
| System | Key Features | Advantages | Disadvantages | Ideal Use Case |
|---|---|---|---|---|
| One-Plasmid System [49] | Cas9 and gRNA encoded on a single vector (e.g., Lenti-Cas9-gRNA-GFP). | Simplified workflow; reduced experimental variables. | Large vector size may reduce viral titer; transient expression can limit editing efficiency. | Generating a single knockout in a easy-to-transfect cell line. |
| Two-Plasmid System [49] | Cas9 expressed from a stable cell line or one plasmid, gRNA from a separate vector (e.g., LentiVCas9puro + LRG2.1). | Enables creation of a reusable Cas9-expressing parental line; allows for multiplexing with different gRNAs. | Constitutive Cas9 expression may increase off-target effects. | Generating multiple knockouts in the same cell line background. |
| Ribonucleoprotein (RNP) [49] | Direct delivery of pre-complexed Cas9 protein and gRNA. | Highest editing efficiency; reduced off-target effects and cytotoxicity; fastest action. | Requires purification or purchase of Cas9 protein; may be cost-prohibitive for large scales. | Hard-to-transfect cells (e.g., primary cells) or when minimal off-targets are critical. |
A critical subsequent step is the design of the guide RNA (gRNA). To maximize the probability of a complete knockout, adhere to the following heuristics:
- Target Functional Domains: Prioritize gRNAs that target exons encoding critical protein domains (e.g., catalytic sites, DNA-binding domains) to ensure indels disrupt function [49].
- Employ a Multi-Guide Strategy: Using two gRNAs targeting the same gene can delete a large genomic segment, dramatically increasing the likelihood of a null allele and avoiding escape mechanisms like alternative splicing or translational re-initiation [49].
- Leverage Bioinformatics Tools: Utilize specialized software (e.g., Benchling, GUIDES) to select gRNAs with high on-target scores and minimal predicted off-target activity in the genome of your specific cell line [49] [8].
- Design Control gRNAs: Always include gRNAs targeting "safe harbor" loci like Rosa26 or AAVS1 to generate control clonal lines for downstream experiments [49].
Detailed Experimental Protocols
Protocol 1: Transfection of CRISPR Constructs
This protocol outlines a method for transient transfection using polyethylenimine (PEI), a cost-effective and highly efficient reagent [50] [51], though lipid-based reagents like Lipofectamine are also widely used.
Materials & Reagents
- Plasmid DNA: All-in-one Cas9-gRNA plasmid or separate Cas9 and gRNA plasmids.
- Polyethylenimine (PEI), linear 25 kDa or 40 kDa [52] [51].
- Opti-MEM or other serum-free medium.
- Target cells (e.g., HEK 293T, HCT-116).
- Complete cell culture medium.
Procedure
- Seed Cells: One day before transfection, seed cells in a culture plate to achieve 60-80% confluency at the time of transfection. Increasing cell seeding density can significantly improve transfection outcomes [51].
- Prepare Complexes:
- For one well of a 6-well plate, dilute 2.5 µg of total plasmid DNA in 150 µL of Opti-MEM.
- In a separate tube, dilute PEI in 150 µL of Opti-MEM at an N/P ratio of 8.0. Studies show this ratio often achieves optimal transfection efficiency for PEI/DNA nanoparticles [51].
- Rapidly mix the PEI solution with the DNA solution by pipetting. Vortexing is not recommended.
- Incubate the mixture at room temperature for 15-20 minutes to allow stable polyplex formation.
- Transfect Cells: Add the DNA-PEI complex mixture dropwise to the cells. Gently swirl the plate to ensure even distribution.
- Post-Transfection Incubation:
- Assay or Select: Analyze transfection efficiency via fluorescence 24-48 hours post-transfection if using a reporter. Begin antibiotic selection (e.g., puromycin) if applicable, 24-48 hours post-transfection.
Transfection Optimization Notes:
- Reagent Selection: Performance is cell-line dependent. Systematic comparisons show that while Lipofectamine 2000 can achieve high efficiency, it is often associated with significant cytotoxicity. In-house cationic lipids (e.g., DOTAP/DOPE formulations) and PEI can offer a more cost-effective alternative with high mRNA transfection efficiency and lower cytotoxicity [52]. JetPrime is another commercial reagent noted for high, though sometimes cytotoxic, efficiency [50].
- Parameter Tuning: For difficult-to-transfect cells like T cells, reversing the protocol (adding cells to pre-dispensed complexes in vials) and modifying cellular physiology with hypotonic extracellular media at pH 9.0 have been shown to dramatically enhance uptake and efficiency [51].
Protocol 2: Single-Cell Isolation by Limiting Dilution
The goal is to derive a homogeneous cell population from a single progenitor. While fluorescence-activated cell sorting (FACS) is an alternative, limiting dilution is accessible and effective.
Materials & Reagents
- Transfected and selected cell pool.
- Conditioned medium: Filtered supernatant from a confluent culture of the same cell line.
- 96-well flat-bottom tissue culture plates.
- Complete growth medium.
Procedure
- Prepare Cells: Harvest the transfected and selected cell pool. Perform a accurate cell count using a hemocytometer or automated counter.
- Serially Dilute Cells: Prepare a series of cell suspensions in complete growth medium supplemented with 20-30% conditioned medium. The conditioned medium provides essential growth factors and improves single-cell survival [8].
- Dilution A: 10-20 cells/mL
- Dilution B: 5-10 cells/mL
- Dilution C: 1-5 cells/mL
- Plate Cells: Dispense 100 µL of each cell suspension into the wells of 96-well plates (i.e., resulting in 1-2, 0.5-1, and 0.1-0.5 cells/well, respectively). For the most dilute suspension, plating a larger number of plates is recommended.
- Incubate and Monitor:
- Place plates in a humidified 37°C, 5% CO₂ incubator.
- After 24 hours, carefully inspect each well under a microscope and mark wells that received exactly one cell. Re-inspect over the next few days to confirm clonality. Any well with more than one cell at the start should be excluded from further analysis [8].
- Expand Clones: Once a single cell has proliferated to cover ~30-50% of the well's surface, carefully trypsinize and transfer it to a well of a 24-well plate, and subsequently to a 12-well plate, 6-well plate, and finally a T25 flask. Always use conditioned medium during the initial expansion phases to support growth.
Protocol 3: Clonal Expansion and Validation
This phase ensures the viability and verifies the genotype of the isolated clones.
Materials & Reagents
- Expanded clonal cell lines.
- Lysis buffer for genomic DNA extraction.
- PCR reagents.
- Western blotting equipment and antibodies against the target protein.
- Sanger or Next-Generation Sequencing (NGS) services.
Procedure
- Maintain Clonal Cultures: Continue passaging the expanding clonal lines, cryopreserving aliquots at early passages (e.g., upon reaching a T25 flask). This preserves the clone and provides a backup.
- Validate Genomic Edits:
- Genomic DNA Extraction: Harvest cells from a confluent well of a 24- or 12-well plate to extract genomic DNA.
- PCR Amplification: Design primers flanking the CRISPR target site and amplify the region.
- Sequence Analysis: Submit the PCR product for Sanger sequencing. For a more comprehensive view of the editing spectrum, especially in potentially mixed populations, NGS is recommended [49] [14]. Analyze the sequencing traces for frameshift indels or large deletions in both alleles.
- Confirm Protein Knockout: The definitive validation is the absence of the target protein.
- Western Blotting: Lyse a portion of the cells and perform a Western blot with an antibody specific for the protein of interest. A true knockout should show a complete absence of the protein band. Mass spectrometry can also provide confirmation at the proteomic level [14].
- Functional Validation: Depending on the gene's known function, perform relevant assays (e.g., proliferation assays, immunofluorescence, or flow cytometry) to confirm the expected phenotypic change.
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for CRISPR Knockout Generation
| Item | Function/Explanation | Example Products/Catalog Numbers |
|---|---|---|
| CRISPR Plasmids | Vectors for expressing Cas9 nuclease and guide RNA(s). | Lenti-Cas9-gRNA-GFP (Addgene #124770), LentiVCas9puro (Addgene #108100), pSpCas9(BB)-2A-Puro (PX459, Addgene #48139) [49] [8]. |
| Transfection Reagents | Chemical agents that form complexes with nucleic acids to facilitate cellular uptake. | Linear PEI (25kDa, 40kDa) [52] [51], Lipofectamine 3000 [8], JetPrime [50], in-house cationic lipids (e.g., DOTAP/DOPE) [52]. |
| Selection Antibiotics | To select for cells that have successfully incorporated the CRISPR plasmid. | Puromycin, Geneticin (G418). The optimal concentration must be determined empirically via a kill curve assay (e.g., MTT assay) [8]. |
| Conditioned Medium | Spent medium from a confluent culture, rich in growth factors that support single-cell survival. | Prepared in-lab from the parental cell line. |
| Cell Strainer Mesh | To ensure a single-cell suspension before limiting dilution, preventing clumping. | 40 µm nylon mesh filters [8]. |
| Validation Antibodies | For confirming the absence of the target protein via Western blot or immunocytochemistry. | Target-specific antibodies from various suppliers. |
Workflow and Data Analysis Visualization
CRISPR Knockout Generation Workflow
Single-Cell to Clonal Line Process
The generation of knockout cell lines using CRISPR-Cas9 has become a fundamental technique in biomedical research. However, achieving efficient and precise genetic modifications in challenging cell types, including induced pluripotent stem cells (iPSCs) and primary cells, presents unique technical hurdles. These difficulties are compounded when attempting multiplexed genome editing—simultaneously targeting multiple genomic loci—which is essential for modeling complex diseases and functional genomics screens. This application note details optimized protocols and strategic approaches to overcome these challenges, enabling robust knockout generation in even the most recalcitrant cell types.
A primary obstacle in iPSC gene editing is the frequent silencing of Cas9 expression, particularly during directed differentiation, even when integrated into well-characterized safe harbor loci like AAVS1 [53]. In primary and postmitotic cells, limitations include low transfection efficiency, heightened sensitivity to DNA damage, and fundamentally different DNA repair kinetics compared to immortalized cell lines [54]. Simultaneously, multiplexed editing demands careful orchestration of multiple guide RNAs (gRNAs) and strategies to mitigate cellular toxicity from concurrent double-strand breaks (DSBs) [55]. The protocols herein are designed to address these specific bottlenecks.
Overcoming Cas9 Silencing in iPSCs
A significant challenge in using engineered iPSC lines is the progressive silencing of transgenes like Cas9. Research demonstrates that inserting the Cas9-EGFP construct into the exon 9 of the GAPDH gene using Selection by Essential Gene Exon Knock-in (SLEEK) technology can effectively bypass this silencing [53]. This strategy leverages the strong, constitutive expression from the endogenous GAPDH promoter.
Protocol: Generating iPSCs with Stable Cas9-EGFP Expression via SLEEK Technology
This protocol outlines the steps for creating iPSC lines with sustained Cas9 expression by targeting the GAPDH locus [53].
- Step 1: Plasmid Construction. Generate a donor plasmid containing a Cas9-EGFP-T2A-recoded GAPDH exon 9 cassette flanked by homology arms specific to the GAPDH locus. The recoded exon preserves the wild-type GAPDH amino acid sequence but alters the DNA sequence to prevent re-cleavage by Cas9 after successful knock-in.
- Step 2: gRNA Design. Design a gRNA to induce a DSB within the endogenous GAPDH exon 9.
- Step 3: iPSC Electroporation. Co-electroporate the donor plasmid and gRNA/Cas9 complex (as ribonucleoprotein, RNP) into iPSCs. The RNP format enhances editing efficiency and reduces off-target effects.
- Step 4: Selection and Clonal Isolation. Unlike antibiotic selection, this protocol uses negative selection based on cell viability. Only cells that have undergone successful homology-directed repair (HDR) restore a functional GAPDH gene and survive. Cells that repair the DSB via non-homologous end joining (NHEJ) harbor disruptive mutations and fail to proliferate due to loss of GAPDH function. Surviving cells are clonally expanded.
- Step 5: Validation. Validate knock-in clones using a PCR-based strategy with primers located outside both the 5' and 3' homology arms, followed by Sanger sequencing. Confirm sustained Cas9 expression and function, especially after differentiation.
Quantitative Data on Optimized iPSC Knockout
Systematic optimization of parameters in a doxycycline-inducible Cas9 (iCas9) iPSC system has demonstrated remarkable knockout efficiencies, as summarized in Table 1 [12].
Table 1: High-Efficiency Knockout in Optimized iPSC Systems
| Editing Type | Target | Optimized INDEL Efficiency | Key Optimization Parameters |
|---|---|---|---|
| Single-Gene Knockout | Various genes | 82% - 93% | Cas9 mRNA/sgRNA RNP delivery; high cell viability post-nucleofection; chemically modified sgRNAs [12]. |
| Double-Gene Knockout | Two distinct genes | > 80% | Co-delivery of two sgRNAs at a 1:1 ratio [12]. |
| Large Fragment Deletion | Two sites in one gene | Up to 37.5% (homozygous) | Use of two sgRNAs flanking the region to be deleted [12]. |
Strategies for Multiplexed Genome Editing
Multiplexed CRISPR-Cas9 editing allows for the simultaneous disruption of multiple genes or the creation of large genomic deletions, which is invaluable for studying gene networks and non-coding elements [55].
Applications of Multiplexed Editing
- Combinatorial Gene Knockout: Enables the study of synthetic lethality and gene interactions. Genome-wide paired gRNA libraries (e.g., CDKO library) can screen for synergistic genetic interactions [55].
- Large Structural Variants: Using two gRNAs to target distant sites within a chromosome can generate large deletions, inversions, or duplications, mimicking structural variations found in human diseases [55].
- Excision of Genomic Elements: Efficiently remove large DNA fragments, such as selectable marker genes from transgenic plants or animals, by employing multiple gRNAs targeting the flanks of the cassette [56].
Protocol: Multiplexed Knockout Using Synthetic crRNAs
This protocol utilizes synthetic CRISPR RNAs (crRNAs) complexed with tracrRNA and Cas9 (as mRNA or protein) for highly efficient multiplexed editing without the need for custom plasmid construction [57].
- Step 1: crRNA Design. Select algorithm-optimized crRNAs for each target gene. Using 2-4 crRNAs per reaction is typical.
- Step 2: RNP Complex Formation. For each target, complex the respective crRNA with tracrRNA to form a guide RNA (gRNA). Then, combine all gRNAs with Cas9 protein (or mRNA) to form the multiplex RNP complex. Some protocols recommend complexing each gRNA separately before combining them to ensure equal representation.
- Step 3: Cell Transfection/Electroporation. Deliver the multiplex RNP complex into cells, preferably a Cas9-stable cell line for highest efficiency. Use a transfection reagent compatible with RNA (e.g., DharmaFECT Duo) or electroporation.
- Step 4: Analysis and Clonal Isolation. After 48-72 hours, analyze the bulk cell population for editing efficiency at all target sites using a mismatch detection assay (e.g., T7E1) or sequencing. Subsequently, isolate single-cell clones and expand them for genotyping and functional validation.
Efficiency of Multiplexed Knockout
The efficiency of generating clones with all target alleles edited varies by cell line and Cas9 delivery method, as shown in Table 2 [57].
Table 2: Efficiency of Three-Gene Multiplexed Knockout in Different Systems
| Cell Type | Cas9 Source | Clonal Lines with 3/3 Genes Edited | Clonal Lines with All Alleles of 3 Genes Edited |
|---|---|---|---|
| HEK293T | Stable Expression | 31% | 12% |
| HEK293T | mRNA | 25% | 6% |
| U2OS | Stable Expression | 100% | 58% |
| U2OS | Protein (RNP) | 25% | 15% |
Editing in Primary and Postmitotic Cells
CRISPR editing outcomes are profoundly different in non-dividing cells, such as neurons and cardiomyocytes, compared to dividing cells [54]. These cells predominantly employ the non-homologous end joining (NHEJ) pathway and show negligible homology-directed repair (HDR) activity due to their exit from the cell cycle.
Key Challenges and Findings
- Prolonged Repair Kinetics: DSB repair in postmitotic human neurons is slow, with indels continuing to accumulate for up to two weeks after Cas9 delivery, unlike in iPSCs where repair plateaus within days [54]. This necessitates extended culture periods post-editing before analysis.
- Distinct Repair Outcome Bias: Neurons favor small indels and unedited outcomes associated with classical NHEJ, while isogenic iPSCs show a higher proportion of microhomology-mediated end joining (MMEJ) outcomes, which are larger deletions [54].
- Delivery Challenges: Standard transfection methods are often inefficient or toxic. Virus-like particles (VLPs) pseudotyped with VSVG and/or BaEVRless (BRL) envelopes can achieve >97% delivery efficiency of Cas9 RNP into human iPSC-derived neurons [54].
Protocol: CRISPR Editing in Human iPSC-Derived Neurons
- Step 1: Differentiate iPSCs into Postmitotic Neurons. Use a validated protocol to generate cortical excitatory neurons. Confirm postmitotic status by Ki67 negativity (>99%) and neuronal markers (e.g., ~95% NeuN positive) [54].
- Step 2: Deliver Cas9 via VLPs. Produce VSVG/BRL-co-pseudotyped Friend Murine Leukemia Virus (FMLV) VLPs loaded with Cas9-sgRNA RNP complexes. Transduce the neuronal culture with these VLPs.
- Step 3: Long-Term Culture and Analysis. Maintain the transduced neurons in culture for at least 14-16 days to allow for the full accumulation of indel mutations. Harvest genomic DNA and analyze editing outcomes by deep sequencing.
The Scientist's Toolkit: Essential Reagents and Solutions
Successful editing in difficult cells requires a carefully selected set of tools and reagents. Table 5 outlines key solutions for advanced CRISPR applications.
Table 5: Research Reagent Solutions for Advanced Knockout Generation
| Reagent / Solution | Function | Application Context |
|---|---|---|
| SLEEK Knock-in System | Inserts transgene into exon 9 of GAPDH to prevent epigenetic silencing. | Creating iPSC lines with sustained, constitutive Cas9 expression [53]. |
| Chemically Modified sgRNA | 2'-O-methyl-3'-thiophosphonoacetate modifications enhance nuclease resistance and stability. | Improves editing efficiency in all cell types, particularly sensitive iPSCs and primary cells [12]. |
| Synthetic crRNA/tracrRNA | Pre-designed, synthetic guide RNA components for RNP complex formation. | Enables rapid, DNA-free multiplexed editing without cloning [57]. |
| Virus-Like Particles (VLPs) | Engineered particles (e.g., FMLV-, HIV-based) for protein cargo delivery. | Highly efficient delivery of Cas9 RNP into hard-to-transfect primary and postmitotic cells [54]. |
| Inducible Cas9 (iCas9) | Doxycycline-regulated Cas9 expression system. | Allows control over timing of nuclease activity, improving cell health and enabling study of essential genes [12]. |
Workflow Visualization
The following diagram illustrates the strategic decision-making process and core workflows for generating knockouts in difficult cell types, integrating the key protocols discussed in this note.
Advanced CRISPR applications in iPSCs, primary cells, and for multiplexed engineering are now achievable with robust efficiency by adopting tailored strategies. Key to success is overcoming delivery barriers and adapting to the unique cellular physiology of each system—whether by preventing transgene silencing in iPSCs, exploiting synthetic RNA complexes for multiplexing, or utilizing specialized delivery vehicles like VLPs for neurons. The protocols and data summarized here provide a framework for researchers to design and execute sophisticated gene knockout experiments, accelerating the development of complex disease models and paving the way for next-generation cell therapies.
Solving Common CRISPR Challenges: A Troubleshooting Guide for Low Efficiency and Off-Target Effects
The generation of robust CRISPR-Cas9 knockout cell lines represents a cornerstone technique in modern molecular biology, enabling functional studies of genetic loss-of-function in disease modeling and therapeutic development. Despite the widespread adoption of CRISPR-Cas9 technology, many research groups encounter significant challenges in achieving consistent high knockout efficiency, leading to unreliable data, wasted resources, and delayed project timelines. Low knockout efficiency manifests as heterogeneous editing outcomes within cell populations, residual protein expression despite detected genetic alterations, and ultimately, an inability to draw meaningful biological conclusions from functional assays.
The fundamental challenge stems from the complex interplay between multiple experimental variables, including sgRNA design quality, cellular delivery efficiency, DNA repair machinery activity, and the specific biological context of the target cell line. This application note provides a systematic framework for diagnosing the root causes of low knockout efficiency and implementing proven solutions to overcome these limitations, with all protocols and methodologies framed within the context of a broader thesis on CRISPR-Cas9 knockout cell line generation research.
Diagnostic Framework: Identifying the Causes of Low Efficiency
Before implementing corrective measures, researchers must first accurately diagnose the underlying causes of poor knockout performance. A systematic approach to diagnosis prevents wasted effort on irrelevant optimizations and directs attention to the most impactful parameters.
Efficiency Assessment Methods and Their Limitations
Accurately quantifying knockout efficiency presents its own challenges, as different detection methods provide varying levels of insight and come with distinct limitations.
Table 1: Comparison of Knockout Efficiency Assessment Methods
| Method | Information Provided | Key Limitations | Optimal Use Case |
|---|---|---|---|
| Sanger Sequencing + Decomposition (ICE, TIDE) | Quantifies INDEL percentage and spectrum | Cannot detect large deletions; limited multiplexing capability | Rapid assessment of editing efficiency at single target sites [12] |
| Next-Generation Sequencing | Comprehensive characterization of all editing events; highly quantitative | Higher cost and computational burden; overkill for initial screening | Gold standard for final validation; complex editing analysis [58] |
| Western Blotting | Direct confirmation of protein loss; functional validation | Cannot distinguish heterozygous from homozygous knockouts; antibody quality dependent | Essential validation step to confirm functional knockout [58] [12] |
| qPCR | mRNA expression levels | Poor correlation with functional knockout; misses frameshifts without NMD | Limited utility for knockout validation; not recommended as primary method [59] |
A critical consideration in efficiency assessment is the potential disconnect between genetic and functional knockout. Research has demonstrated that edited cell pools can exhibit INDEL efficiencies exceeding 80% at the DNA level while retaining target protein expression, as evidenced by a case study targeting exon 2 of ACE2 [12]. This underscores the necessity of multi-modal validation, particularly incorporating protein-level detection.
Systematic Diagnostic Workflow
The following diagnostic pathway provides a logical framework for identifying the specific factors limiting knockout efficiency in a given experimental system:
Key Optimization Strategies for Enhanced Knockout Efficiency
sgRNA Design and Selection
sgRNA design represents the most critical parameter influencing knockout efficiency. Beyond basic design principles, several advanced strategies can significantly enhance performance:
Algorithm Selection and Validation
Recent benchmark comparisons of publicly available genome-wide sgRNA libraries have revealed substantial differences in the predictive performance of various scoring algorithms. The Vienna Bioactivity CRISPR (VBC) score has demonstrated superior performance in predicting sgRNA efficacy, with guides selected using this metric showing stronger depletion of essential genes in lethality screens compared to other libraries [22]. When designing sgRNAs, researchers should prioritize algorithms with demonstrated predictive power and ideally select multiple sgRNAs per target for empirical testing.
Structural Optimization
The structural configuration of sgRNA itself significantly impacts knockout efficiency. Systematic investigation has revealed that extending the sgRNA:DNA duplex by approximately 5 base pairs and mutating the fourth thymine in the terminal poly-T sequence to cytosine or guanine can dramatically improve knockout efficiency across multiple target sites and cell types [35]. This optimized structure enhances stability and potentially improves Cas9 binding, with particularly pronounced benefits for challenging applications like gene deletion.
Table 2: Optimized sgRNA Design Parameters for Enhanced Knockout Efficiency
| Parameter | Standard Approach | Optimized Approach | Efficiency Improvement |
|---|---|---|---|
| Duplex Length | Shortened (10 bp shorter than native) | Extended by ~5 bp | 1.5- to 3-fold increase in protein-level knockout [35] |
| Terminal Sequence | Continuous T-stretch (transcription terminator) | T→C or T→G mutation at position 4 | Significant improvement in transcription efficiency [35] |
| Algorithm Selection | Variable performance across platforms | VBC scoring algorithm | Strongest essential gene depletion in benchmark studies [22] |
| Chemical Modification | Unmodified in vitro transcripts | 2'-O-methyl-3'-thiophosphonoacetate modifications | Enhanced stability, particularly for synthetic sgRNAs [12] |
Delivery Method Optimization
Efficient delivery of CRISPR components remains a fundamental requirement for successful knockout generation. The choice of delivery method should be tailored to the specific cell type and experimental requirements:
Ribonucleoprotein (RNP) Delivery
The use of preassembled Cas9 ribonucleoprotein complexes has emerged as a particularly efficient strategy, especially in challenging cell types. RNP delivery offers rapid editing with reduced off-target effects and minimal residual activity. In fungal systems such as Scedosporium apiospermum, RNP delivery has enabled efficient gene deletion even in wild-type strains, achieving success rates up to 20% of transformants without requiring prior manipulation of DNA repair pathways [60].
Viral and Non-Viral Delivery Systems
For difficult-to-transfect cell types, viral delivery systems (lentivirus, adenovirus) often provide superior transduction efficiency. However, the development of advanced lipid-based nanoparticles has dramatically improved the efficiency of non-viral delivery. Research demonstrates that lipid-based transfection reagents like DharmaFECT or Lipofectamine 3000 facilitate CRISPR component uptake through enhanced endocytosis, while electroporation serves as a viable alternative for cell lines resistant to lipid-based methods [58].
Cellular and System-Level Optimization
Beyond molecular design and delivery, cellular context significantly influences editing outcomes:
Cas9 Expression Systems
The method of Cas9 expression substantially impacts editing efficiency and reproducibility. Stable Cas9-expressing cell lines eliminate variability associated with transient transfection and provide more consistent editing outcomes. Inducible Cas9 systems offer additional control, with optimized doxycycline-inducible systems achieving remarkable INDEL efficiencies of 82-93% for single-gene knockouts and over 80% for double-gene knockouts in human pluripotent stem cells [12].
DNA Repair Pathway Modulation
The competition between non-homologous end joining (NHEJ) and homology-directed repair (HDR) pathways fundamentally influences knockout outcomes. In wild-type fungal strains with highly efficient non-homologous recombination, successful gene disruption requires prior manipulation of the NHEJ pathway, such as disruption of the KU70 gene [60]. While similar approaches are less common in mammalian cells, understanding the DNA repair propensity of specific cell types can inform experimental design and interpretation.
Integrated Protocol for Generating Knockout Cell Lines
What follows is a detailed, optimized protocol for generating knockout cell lines in adherent cell cultures, incorporating the critical optimization strategies discussed previously.
sgRNA Design and Cloning (Timing: ∼10 days)
- Target Identification: Using the UCSC genome browser, identify the target region and obtain flanking sequences (50-100 bp on each side) [61].
- sgRNA Selection: Design at least two separate pairs of sgRNAs using the CRISPOR tool, prioritizing targets with high VBC scores [22]. For each target, design two sgRNAs facing each other to enable dual cutting and fragment deletion.
- Structural Optimization: Implement extended duplex (+5 bp) and T→C mutation at position 4 in the sgRNA template design [35].
- Cloning into Delivery Vector: Clone optimized sgRNA sequences into the pgRNA-humanized vector (Addgene #44248) using the BstXI and XhoI restriction sites [61].
- Quality Control: Verify clone integrity by Sanger sequencing using the sgRNA sequencing oligo (5'-CCCTGCCCCGGTTAATTTGC-3').
Delivery and Cell Selection (Timing: ∼14 days)
- Virus Production (for lentiviral delivery):
- Co-transfect HEK293FT cells with the transfer plasmid (pgRNA-humanized with sgRNA insert), psPAX2 (packaging plasmid), and pCMV-VSV-G (envelope plasmid) using Lipofectamine 2000 [61].
- Collect viral supernatant at 48 and 72 hours post-transfection, concentrate if necessary, and titer determination.
- Cell Transduction:
- Plate target cells (e.g., HeLa, MCF7) in poly-D-lysine coated dishes to enhance adhesion [61].
- Transduce with viral particles in the presence of 8 μg/mL polybrene via spinfection (2000 × g, 90 minutes, 32°C).
- For RNP delivery, complex purified Cas9 protein with synthetic, chemically modified sgRNAs and deliver via nucleofection using cell type-specific protocols [12].
- Selection and Single-Cell Cloning:
- Begin antibiotic selection (e.g., puromycin, 1-5 μg/mL) 48 hours post-transduction for 5-7 days.
- Harvest cells and perform limiting dilution in 96-well plates to obtain single-cell clones.
- Expand clones for 2-3 weeks with regular medium changes.
Validation and Characterization (Timing: ∼7 days)
- Genomic DNA Extraction:
- Extract genomic DNA using isolation buffer (100 mM NaCl, 10 mM Tris-Cl, 25 mM EDTA, 0.5% SDS) with freshly added proteinase K (0.1 mg/mL) [61].
- Incubate at 55°C for 4 hours or overnight, followed by phenol:chloroform:isoamyl alcohol extraction and ethanol precipitation.
- Primary PCR Screening:
- Design "PCRout" primers flanking the target deletion region (amplicon size: 1-3 kb for large deletions).
- Perform PCR amplification using high-fidelity DNA polymerase (e.g., KOD Hot Start).
- Identify potential knockout clones by detecting size changes on agarose gel electrophoresis.
- Secondary Validation:
- For clones showing deletion patterns, perform nested "PCRin" with primers inside the deleted region (no product expected in successful knockouts).
- Confirm exact editing by Sanger sequencing of PCR products and decomposition analysis using ICE or TIDE algorithms [12].
- Functional Validation:
- Confirm protein loss by Western blotting using validated antibodies.
- For critical applications, perform additional functional assays specific to the target gene (e.g., enzymatic assays, substrate accumulation tests).
Essential Research Reagents and Tools
Table 3: Key Research Reagent Solutions for CRISPR Knockout Generation
| Reagent Category | Specific Examples | Function and Application Notes |
|---|---|---|
| Cas9 Expression Systems | lentiCas9-Blast (Addgene #52962), Dox-inducible iCas9 systems | Provides Cas9 nuclease; inducible systems enable temporal control and higher efficiency [12] [61] |
| sgRNA Cloning Vectors | pgRNA-humanized (Addgene #44248) | sgRNA expression backbone with human U6 promoter [61] |
| Delivery Reagents | Lipofectamine 2000, DharmaFECT, Lipid nanoparticles (LNPs) | Chemical transfection; optimal for adherent cell lines [58] |
| Viral Packaging System | psPAX2 (Addgene #12260), pCMV-VSV-G (Addgene #8454) | Lentiviral packaging for difficult-to-transfect cells [61] |
| Validation Tools | ICE (Synthego), TIDE decomposition, Anti-target protein antibodies | Editing efficiency quantification and functional knockout confirmation [12] |
| Cell Culture Supplements | Poly-D-lysine, Puromycin, Polybrene | Enhances adhesion, selection, and transduction efficiency [61] |
Achieving consistent high-efficiency knockout in cell lines requires a systematic approach that addresses the multiple interdependent factors influencing editing outcomes. By implementing optimized sgRNA designs with extended duplexes and terminal modifications, selecting appropriate delivery methods matched to cell type capabilities, utilizing stable or inducible Cas9 expression systems, and employing comprehensive multi-modal validation strategies, researchers can significantly improve the success and reliability of their knockout cell line generation efforts. The protocols and frameworks presented herein provide a roadmap for troubleshooting inefficient editing and establishing robust, reproducible workflows for CRISPR-based functional genomics within the broader context of gene editing research and therapeutic development.
As CRISPR technology continues to evolve, emerging approaches such as dual-targeting sgRNA strategies [22] and novel Cas variants with enhanced specificity promise to further improve knockout efficiency and expand the range of accessible genomic targets.
Optimizing Transfection Efficiency for Challenging Cell Lines
Within the broader scope of CRISPR-Cas9 knockout cell line generation research, optimizing transfection efficiency represents a critical bottleneck, particularly when working with challenging cell types. Successful genome editing hinges on the efficient delivery of CRISPR components into the nucleus of living cells while maintaining high cell viability [14]. Challenging cell lines—including primary cells, stem cells, and immune cells such as THP-1—often exhibit low transfection efficiency and increased sensitivity to transfection-induced cytotoxicity [41] [14]. This application note provides a comprehensive guide to established and emerging strategies for optimizing transfection in these difficult contexts, complete with detailed protocols and analytical frameworks for researchers and drug development professionals.
Understanding Transfection Challenges in Different Cell Types
The term "challenging cell lines" encompasses a range of cell types with distinct biological properties that complicate standard transfection protocols. Primary cells, derived directly from living tissue, have limited propagation capacity and fewer cell divisions, reducing opportunities for CRISPR components to enter the nucleus [41]. Stem cells (including ES and iPS cells) are sensitive to manipulation and require careful maintenance of viability and pluripotency during transfection [62] [41]. Suspension immune cells like THP-1 and Jurkat are particularly difficult to transfect using traditional methods due to their non-adherent nature and specialized cell membranes [13] [63].
A survey of CRISPR researchers reveals that transfection condition optimization is considered the most difficult step in the workflow by many scientists, with the average researcher requiring nearly five months to generate a CRISPR knockout cell line and often restarting experiments three to four times before success [14]. The biological underpinnings of these challenges include altered cell repair mechanisms, lower baseline viability, and inefficient nuclear entry of CRISPR components in non-dividing cells [14].
Strategic Approaches to Transfection Optimization
Selection of Transfection Method
The choice of transfection method should be guided by cell type, CRISPR component format, and experimental requirements. The table below compares the primary transfection methods used for challenging cell lines:
Table 1: Comparison of Transfection Methods for Challenging Cell Lines
| Method | Principle | Advantages | Disadvantages | Best For |
|---|---|---|---|---|
| Lipofection [64] | Lipid complexes fuse with cell membrane | Cost-effective, high throughput, easy operation | Less efficient for sensitive cells, serum interference | Common cell lines with low cytotoxicity requirements |
| Electroporation [41] [64] | Electrical pulses form temporary pores in membrane | Broad cell type applicability, high efficiency | High cell toxicity, requires parameter optimization | High-efficiency delivery in suspension cells |
| Nucleofection [62] [41] | Electroporation optimized for nuclear delivery | Direct nuclear transfer, high efficiency for primary cells | Specialized equipment required | Primary cells, stem cells, hard-to-transfect lines |
| Lentiviral Delivery [13] [64] | Viral transduction | High efficiency, works with difficult-to-transfect cells | Time-consuming, biosafety concerns, size limitations | Stable cell line generation, immune cells |
For challenging cell lines like THP-1, lentiviral delivery often provides superior results. One protocol demonstrated successful generation of single-gene knockouts in hard-to-transfect THP-1 cell lines using CRISPR/Cas9 delivered via lentivirus, achieving high efficiency where other methods failed [13].
CRISPR Component Format Selection
The format of CRISPR components significantly influences transfection success:
- Plasmid DNA: Requires nuclear entry, transcription, and translation; higher off-target risk [41]
- mRNA: Faster protein expression but susceptible to degradation [41]
- Ribonucleoprotein (RNP) Complexes: Pre-complexed Cas9 protein and guide RNA; rapid activity, reduced off-target effects, and improved editing efficiency in hard-to-transfect cells [65] [41]
RNP complexes are particularly advantageous for challenging cell types as they bypass transcription and translation requirements, immediately engaging in genome editing upon nuclear delivery [41].
Detailed Optimization Protocols
Lentiviral Transfection Protocol for THP-1 Cells
This protocol adapts the method described by Srivastava et al. for generating single-gene knockouts in hard-to-transfect THP-1 cell lines [13]:
Day 1: sgRNA Design and Vector Preparation
- Design specific sgRNAs using bioinformatics tools to minimize off-target effects
- Clone sgRNA sequence into a CRISPR lentiviral vector (e.g., lentiCRISPR v2)
- Transform into competent bacteria and select with appropriate antibiotics
- Isolate plasmid DNA using high-purity endotoxin-free kit
Day 2-4: Viral Production
- Plate HEK293T packaging cells in complete medium
- Co-transfect with CRISPR transfer plasmid and packaging plasmids (psPAX2, pMD2.G) using PEI transfection reagent
- After 6 hours, replace transfection medium with fresh complete medium
- Collect viral supernatant at 48 and 72 hours post-transfection
- Concentrate viral particles using ultracentrifugation or PEG-it virus precipitation solution
- Titrate virus using appropriate method (e.g., p24 ELISA, functional titer)
Day 5: Cell Preparation and Transduction
- Culture THP-1 cells in RPMI-1640 with 10% FBS to density of 5×10^5 cells/mL
- Spinulate cells (centrifuge at 500 × g and resuspend in fresh medium)
- Add concentrated lentivirus at appropriate MOI (determined by pilot optimization) in the presence of 8 μg/mL polybrene
- Centrifuge at 800 × g for 30 minutes at 32°C (spinoculation) to enhance infection
- Incubate at 37°C, 5% CO2 for 24 hours
Day 6: Selection and Expansion
- Replace medium with fresh complete medium containing appropriate selection antibiotic (e.g., 1-2 μg/mL puromycin)
- Maintain selection for 5-7 days, monitoring for control cell death
- Expand surviving polyclonal population for validation
High-Throughput Optimization Using Fluorescent Reporters
For systematic optimization across multiple parameters, implement a reporter-based quantification system:
Stable Reporter Cell Line Development [65]
- Engineer a dual-fluorescence (RFP-GFP) reporter construct with a target sequence between the two fluorophores
- Stably transfect into your target cell line and sort for high RFP expression
- When functional CRISPR components are delivered, indels at the target site can restore GFP expression
- Use flow cytometry or microplate reading to quantify GFP+/RFP+ cells as a measure of editing efficiency
Microplate Reader-Based Quantification [65]
- Seed stable reporter cells in 96-well plates at defined densities (e.g., 10,000-50,000 cells/well)
- Transfert with CRISPR components using different optimization parameters
- After 72-96 hours, measure RFP and GFP fluorescence using appropriate filter sets
- Calculate GFP/RFP fluorescence ratio and compare to standard curve to determine editing efficiency
Table 2: Transfection Optimization Matrix for Systematic Testing
| Parameter | Test Range | Measurement |
|---|---|---|
| Cell Density | 50-90% confluency (adherent); 5×10^5-2×10^6 cells/mL (suspension) [66] | Editing efficiency, viability |
| DNA:Reagent Ratio | 1:1 to 6:1 (varies by reagent) [63] | Transfection efficiency, toxicity |
| CRISPR Format | Plasmid, mRNA, RNP [41] | Editing efficiency, off-target rate |
| Incubation Time | 4-72 hours post-transfection | Efficiency, viability |
| Serum Condition | Serum-free vs. serum-containing [66] | Complex formation, efficiency |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Research Reagent Solutions for Transfection Optimization
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Lipofectamine 3000 [63] | Lipid-based nucleic acid delivery | Optimal for many immortalized cell lines; lower toxicity than Lipofectamine 2000 |
| ViaFect [63] | Lipid-based transfection reagent | Low toxicity, works in presence of serum; suitable for a broad range of cell lines |
| FuGENE HD [63] | Non-liposomal transfection reagent | Low cytotoxicity, works in presence of serum; good for sensitive cells |
| Hieff Trans [64] | Liposomal transfection reagent | Suitable for suspension cells and difficult-to-transfect primary cells; works in serum |
| Polyethylenimine (PEI) [64] | Cationic polymer for nucleic acid complexation | Cost-effective for large-scale transfections; optimal for HEK293 and CHO cells |
| pmaxGFP Vector [62] | Positive control for optimization | Validates transfection efficiency independent of editing efficiency |
| CellTiter-Fluor [63] | Cell viability assay | Measures viability post-transfection without cell lysis |
| Dual-fluorescence Reporter System [65] | Quantification of functional CRISPR uptake | Measures nuclease activity rather than just transfection |
Validation and Troubleshooting
Validation of Successful Knockout
A multi-level validation approach ensures accurate confirmation of gene knockout:
- Genomic Level: Sanger sequencing or next-generation sequencing to verify indels at the target locus [14]
- Transcript Level: qPCR to confirm absence of target gene mRNA [14]
- Protein Level: Western blot or flow cytometry to verify loss of target protein expression [13] [14]
- Functional Level: Phenotypic assays relevant to the target gene's function
Troubleshooting Common Issues
Low Transfection Efficiency
- Verify nucleic acid quality and quantity
- Optimize cell density at transfection
- Test different transfection reagents/methods
- Include positive control (e.g., pmaxGFP) [62]
High Cell Death
- Reduce reagent:DNA ratio
- Shorten exposure time to transfection complexes
- Use lower cytotoxicity reagents (e.g., ViaFect, FuGENE HD) [63]
- Ensure cells are healthy and at low passage number before transfection [66]
Poor Editing Despite Good Transfection
- Switch to RNP format for faster nuclear activity [41]
- Include nuclear localization signals if using DNA formats
- Validate sgRNA activity using in vitro cleavage assay
- Test multiple sgRNAs against the same target
Workflow Visualization
Diagram 1: Transfection Optimization Workflow - This diagram illustrates the systematic approach to optimizing transfection conditions for challenging cell lines, with iterative refinement based on validation results.
Diagram 2: Multi-level Knockout Validation - This validation framework ensures comprehensive confirmation of successful gene knockout across genomic, transcript, protein, and functional levels.
Optimizing transfection efficiency for challenging cell lines requires a systematic approach that balances editing efficiency with cell viability. The protocols and strategies outlined here provide a roadmap for researchers engaged in CRISPR-Cas9 knockout cell line generation. By carefully selecting transfection methods based on cell type characteristics, utilizing appropriate CRISPR component formats, implementing rigorous validation, and applying systematic optimization approaches, researchers can significantly improve success rates even with the most challenging cell models. As CRISPR technology continues to advance, these optimization principles will remain fundamental to generating reliable knockout cell lines for functional genomics and drug development applications.
Strategies to Minimize Off-Target Effects Using High-Fidelity Cas Variants
The generation of CRISPR-Cas9 knockout cell lines is a cornerstone of modern functional genomics and drug discovery research. However, the utility of these cell lines is fundamentally constrained by off-target effects, which can confound phenotypic analyses and compromise experimental validity. Within the context of a broader thesis on CRISPR-Cas9 knockout cell line generation, this application note addresses the strategic implementation of high-fidelity Cas9 variants. These engineered nucleases are designed to minimize non-specific editing while maintaining robust on-target activity, thereby enhancing the reliability of resultant cell lines for downstream applications in target validation and therapeutic development.
Quantitative Comparison of High-Fidelity Cas9 Variants
Systematic evaluation of high-fidelity Cas variants reveals distinct profiles of on-target efficiency and off-target reduction. The data below, synthesized from multiple genomic screens, provides a comparative basis for variant selection.
Table 1: Performance Characteristics of High-Fidelity Cas9 Variants
| Cas9 Variant | Key Mutations | On-Target Efficiency (Relative to WT) | Guidance for Optimal gRNA Design | Primary Applications |
|---|---|---|---|---|
| SpCas9-HF1 | N497A/R661A/Q695A/Q926A [67] | >85% of sgRNAs show >70% activity [67] | Avoid targets with specific sequence features in seed and non-seed regions [68] | Knockout lines requiring maximal specificity [69] |
| HiFi Cas9 | Not specified in sources | Guide-dependent; ~80% of sgRNAs perform well [68] | Sensitive to sequence context at positions 15-18 [68] | Viability screens; high-signal-to-noise applications [68] |
| eSpCas9(1.1) | Not specified in sources | Varies significantly across sgRNAs [70] | Requires careful design with tools like DeepHF [70] | Applications where fidelity is prioritized over maximum efficiency [70] |
| LZ3 Cas9 | Not specified in sources | Correlates with HiFi; similar guide constraints [68] | Shares sequence preferences with HiFi [68] | Often used interchangeably with HiFi in screening [68] |
Experimental Protocol for Knockout Cell Line Generation Using High-Fidelity Variants
gRNA Design and Selection Workflow
The successful application of high-fidelity variants is critically dependent on optimized gRNA design, as these engineered nucleases exhibit distinct sequence preferences compared to wild-type SpCas9 [68] [70].
Procedure:
- Target Site Identification: Identify all potential N20NGG target sites within your gene of interest, considering coding exons and critical functional domains.
- Initial gRNA Filtering: Exclude guides with:
- Low complexity or repetitive sequences
- Homology to multiple genomic loci (assessed via BLAST)
- Overlap with common single nucleotide polymorphisms
- On-target Efficiency Prediction: Utilize specialized prediction tools (e.g., DeepHF) trained specifically on high-fidelity variants rather than wild-type SpCas9 predictors [70].
- Off-target Potential Assessment: Employ multiple algorithms (e.g., Cas-OFFinder, CCTop) to nominate potential off-target sites, prioritizing those with mismatches in the PAM-distal region [71].
- Final Selection: Select 3-5 top-ranking gRNAs based on a balanced consideration of predicted on-target efficiency and off-target risk for empirical testing.
Cell Line Engineering and Validation
This protocol outlines the process for generating knockout cell lines using high-fidelity Cas9 variants, with specific adaptations for their unique properties.
Materials:
- High-fidelity Cas9 expression plasmid (e.g., SpCas9-HF1, HiFi Cas9)
- Validated sgRNA expression construct
- Appropriate cell line (e.g., HEK293T, HCT-116, or other relevant lines)
- Transfection reagent (e.g., Lipofectamine 3000)
- Puromycin or appropriate selection antibiotic
- Lysis buffer for genomic DNA extraction
- PCR reagents
- T7 Endonuclease I or surveyor nuclease
- Agarose gel electrophoresis system
Procedure:
- Cell Seeding: Plate cells at 40-60% confluence in appropriate growth medium 24 hours prior to transfection.
- Transfection Complex Formation:
- For a 6-well plate format, prepare two tubes:
- Tube A: Dilute 2.5 µg Cas9 plasmid + 2.5 µg sgRNA plasmid in 250 µL serum-free medium
- Tube B: Dilute 10-15 µL transfection reagent in 250 µL serum-free medium
- Combine tubes A and B, incubate 15-20 minutes at room temperature
- For a 6-well plate format, prepare two tubes:
- Transfection: Add DNA-lipid complexes dropwise to cells. Gently swirl plate to distribute evenly.
- Selection and Expansion:
- Begin antibiotic selection (e.g., 1-2 µg/mL puromycin) 48 hours post-transfection
- Maintain selection for 5-7 days until control cells (non-transfected) are completely dead
- Expand surviving cells for validation (passage 1, P1)
- On-target Editing Validation:
- Extract genomic DNA from a portion of the pooled population
- PCR amplify the target region (typically 500-800 bp amplicon)
- Perform T7E1 assay:
- Hybridize and reanneal PCR products (95°C for 5 min, ramp down to 25°C at -0.5°C/sec)
- Digest with T7 Endonuclease I for 30 minutes at 37°C
- Analyze fragments by agarose gel electrophoresis
- Calculate indel frequency using band intensity
- Off-target Assessment:
- Select top 3-5 predicted off-target sites per gRNA based on in silico prediction
- Amplify and sequence these loci from the pooled population
- Analyze sequencing traces for indels using computational tools (e.g., ICE analysis [72])
- Single-Cell Cloning:
- After confirming satisfactory on-target editing and minimal off-target effects, serially dilute the pooled population to 0.5-1 cell/100 µL
- Plate 100 µL/well in 96-well plates
- Expand individual clones for 2-3 weeks, monitoring for single-colony formation
- Screen clones for homozygous knockouts via sequencing and functional assays
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for High-Fidelity CRISPR Cell Line Generation
| Reagent Category | Specific Product Examples | Function and Application Notes |
|---|---|---|
| High-Fidelity Cas9 Plasmids | SpCas9-HF1, eSpCas9(1.1), HiFi Cas9, LZ3 Cas9 [68] [67] | Engineered nucleases with reduced non-specific DNA contacts; selection depends on target sequence context [68]. |
| gRNA Design Tools | DeepHF, CRISPOR, Cas-OFFinder [71] [70] | DeepHF is specifically trained on high-fidelity variants and outperforms tools designed for wild-type SpCas9 [70]. |
| Validation Enzymes | T7 Endonuclease I, Surveyor Nuclease | Detect heteroduplex formation in pooled populations; cost-effective but less sensitive than sequencing methods. |
| Off-target Detection | GUIDE-seq, CIRCLE-seq, DISCOVER-seq [71] [72] | Unbiased methods for genome-wide off-target profiling; GUIDE-seq integrates oligos into DSBs for sensitive detection [71]. |
| Analysis Software | ICE (Inference of CRISPR Edits), CRISPResso2 [72] | ICE uses Sanger sequencing traces to quantify editing efficiency and identify off-target effects [72]. |
Mechanistic Basis of High-Fidelity Variants and Safety Considerations
High-fidelity Cas9 variants function through strategic disruption of key protein-DNA interactions, implementing an "excess energy" hypothesis where reduced non-specific binding energy decreases tolerance for mismatched targets [67]. Structural analyses reveal that mutations in the REC3 domain (e.g., in HiFi and LZ3) alter interactions with the RNA/DNA heteroduplex, creating guide-specific efficiency loss that depends on sequence context in both seed and non-seed regions [68].
Critical Safety Considerations:
- Structural Variations: Recent evidence indicates that high-fidelity variants, while reducing point mutation off-targets, still induce substantial on-target structural variations including chromosomal translocations and megabase-scale deletions [73].
- HDR Enhancement Risks: Combining high-fidelity variants with DNA-PKcs inhibitors to enhance HDR efficiency dramatically increases the frequency of chromosomal rearrangements, requiring careful risk-benefit analysis [73].
- Validation Imperative: Comprehensive genomic integrity assessment using methods capable of detecting large structural variations (e.g., CAST-Seq, karyotyping) is essential before considering knockout lines for therapeutic development [73].
Troubleshooting and Optimization Guidelines
- Low On-target Efficiency: If high-fidelity variants show poor activity with selected gRNAs, screen alternative gRNAs with different sequence contexts or consider the Sniper-Cas9 variant which may have different sequence preferences [68].
- Persistent Off-target Effects: For targets with high sequence homology elsewhere in the genome, consider paired nickase strategies or switch to Cas12a systems which have different mismatch tolerance profiles [74].
- Variable Editing Across Clones: When single-cell cloning yields inconsistent knockout efficiency, increase the number of clones screened or use dual-gRNA strategies to delete larger genomic regions for more complete gene disruption.
- Cell Line-Specific Performance: Certain high-fidelity variants may perform differently across cell types; conduct pilot experiments comparing 2-3 variants in your specific cell background before committing to large-scale screening.
Within the broader scope of CRISPR-Cas9 knockout cell line generation research, a critical challenge emerges: the cellular DNA repair machinery does not respond uniformly across different cell types. The same CRISPR-induced double-strand break (DSB) can yield dramatically different mutational outcomes depending on the cellular context, creating significant variability in editing efficiency and clonal survival [54]. This application note examines how cell line-specific DNA repair mechanisms impact CRISPR knockout generation and provides detailed, actionable protocols to overcome these hurdles, enabling more predictable and efficient genome editing outcomes.
DNA Repair Pathway Dynamics Across Cell Types
The fate of a CRISPR-induced DSB is determined by competing DNA repair pathways, each with distinct fidelity and mutational consequences. Understanding which pathways dominate in specific experimental systems is fundamental to predicting and controlling editing outcomes.
Pathway Competition Determines Editing Outcomes
- Non-Homologous End Joining (NHEJ): An error-prone pathway active throughout the cell cycle that directly ligates broken DNA ends, typically resulting in small insertions or deletions (indels) [75] [76].
- Microhomology-Mediated End Joining (MMEJ): An alternative error-prone pathway that utilizes microhomologous sequences for end joining, typically producing larger deletions [54].
- Homologous Recombination (HR): A high-fidelity pathway restricted to S and G2 cell cycle phases that uses a sister chromatid template for precise repair [75].
Cell Type-Specific Repair Heterogeneity
Recent research reveals striking differences in how cells utilize these repair pathways. A 2025 study comparing induced pluripotent stem cells (iPSCs) to genetically identical iPSC-derived neurons found that dividing cells (iPSCs) predominantly use MMEJ, generating larger deletions, while postmitotic cells (neurons) favor NHEJ, producing smaller indels [54]. This fundamental difference in pathway choice directly impacts the distribution of CRISPR editing outcomes, with neurons exhibiting a much narrower range of indels compared to the broad distribution seen in iPSCs [54].
Perhaps more significantly, the kinetics of editing accumulation varies dramatically between cell types. While indels in dividing cells typically plateau within days, postmitotic neurons and cardiomyocytes continue accumulating edits for up to two weeks post-transduction [54]. This prolonged editing timeline has crucial implications for experimental design, particularly regarding the optimal timing for analyzing editing outcomes and isolating clones.
Table 1: DNA Repair Pathway Activity Across Cell Types
| Cell Type | Dominant Repair Pathway | Typical Indel Profile | Repair Kinetics | Therapeutic Implications |
|---|---|---|---|---|
| Dividing Cells (iPSCs, cancer cell lines) | MMEJ > NHEJ | Broad distribution, larger deletions | Fast (plateau in days) | Standard editing protocols generally effective |
| Postmitotic Cells (neurons, cardiomyocytes) | NHEJ > MMEJ | Narrow distribution, smaller indels | Slow (up to 2 weeks) | Requires extended timelines for full editing manifestation |
| HR-Proficient Cells | HR (in S/G2) + NHEJ | Precise repair + indels | Cell cycle-dependent | Lower knockout efficiency; cell cycle synchronization may help |
| HR-Deficient Cells | NHEJ > MMEJ | Mostly indels | Similar to dividing cells | Higher knockout efficiency; sensitive to PARP inhibitors |
Quantitative Analysis of Editing Outcomes
The distribution of CRISPR repair outcomes follows predictable patterns based on dominant repair pathways, enabling researchers to anticipate the mutational landscape in their specific cell models.
Table 2: Quantitative Distribution of CRISPR Repair Outcomes by Cell Type
| Editing Outcome | iPSCs (Dividing) | iPSC-Derived Neurons (Postmitotic) | Primary T Cells (Resting) | HEK293 (Immortalized) |
|---|---|---|---|---|
| No Edit | 5-15% | 20-40% | 15-30% | 5-15% |
| Small Indels (1-10 bp) | 30-50% | 45-65% | 40-60% | 35-55% |
| Large Deletions (>10 bp) | 35-55% | 5-15% | 10-25% | 30-50% |
| Complex Rearrangements | 5-15% | <5% | <10% | 5-15% |
| Insertion:Deletion Ratio | Low (≈0.3) | High (≈0.7) | Moderate (≈0.5) | Low (≈0.4) |
Optimized Experimental Protocols
Protocol 1: CRISPR Knockout in Hard-to-Transfect Suspension Cells
This optimized protocol for THP1 cells demonstrates high efficiency through lentiviral RNP delivery [7].
Background: Suspension immune cells like THP1 present unique challenges for CRISPR editing due to low transfection efficiency and sensitivity to electroporation-induced toxicity. This protocol overcomes these limitations through optimized lentiviral delivery.
Materials & Reagents:
- THP1 cells (ATCC: TIB-202)
- LentiCRISPRv2 vector (Addgene: #52961)
- PsPAX2 packaging plasmid (Addgene: #12260)
- pMD2.G envelope plasmid (Addgene: #12259)
- Polybrene (8 μg/mL)
- Puromycin for selection
- Lenti-X concentrator
- GoStix for viral titer estimation
Procedure:
- sgRNA Design & Cloning (Days 1-6)
- Design sgRNAs using Synthego CRISPR Design Tool targeting exons common to all isoforms [7].
- Synthesize oligos with appropriate overhangs for Golden Gate assembly.
- Clone into BsmBI-digested LentiCRISPRv2 vector using T4 DNA ligase.
- Transform into Stbl3 competent cells and validate through colony PCR and sequencing.
Lentiviral Production (Days 7-10)
- Co-transfect Lenti-X cells with lentiCRISPR-sgRNA, psPAX2, and pMD2.G using Lipofectamine 2000.
- Collect supernatant at 48 and 72 hours post-transfection.
- Concentrate virus using Lenti-X concentrator and estimate titer using GoStix.
Cell Transduction & Selection (Days 11-18)
- Transduce THP1 cells at MOI 5-10 in the presence of 8 μg/mL polybrene.
- Begin puromycin selection (0.5-2 μg/mL) 48 hours post-transduction.
- Maintain selection for 5-7 days until control cells are completely dead.
Validation (Days 19-25)
- Extract genomic DNA from polyclonal population.
- Amplify target region and analyze editing efficiency using TIDE or ICE analysis.
- Isolate single cells by limiting dilution into 96-well plates.
- Expand clones and validate knockout via Western blotting and Sanger sequencing.
Troubleshooting:
- Low viral titer: Ensure 70-80% confluency of Lenti-X cells at transfection.
- Poor transduction efficiency: Optimize polybrane concentration and spinfection parameters.
- Low cell viability during selection: Titrate puromycin concentration using kill curve assay beforehand.
Protocol 2: High-Efficiency Editing in Human Pluripotent Stem Cells
This protocol leverages a doxycycline-inducible Cas9 system to achieve INDEL efficiencies of 82-93% for single-gene knockouts in hPSCs [12].
Background: hPSCs present unique challenges including sensitivity to nucleofection stress and variable editing efficiencies. This protocol systematically optimizes critical parameters to overcome these limitations.
Key Optimizations:
- Use of chemically synthesized, modified sgRNAs with 2'-O-methyl-3'-thiophosphonoacetate modifications at both ends to enhance stability [12].
- Optimized cell-to-sgRNA ratio (8×10⁵ cells with 5μg sgRNA).
- Implementation of repeated nucleofection (3-day interval) for difficult-to-edit loci.
- Doxycycline-inducible Cas9 expression for controlled nuclease activity.
Procedure:
- Cell Preparation
- Culture hPSCs-iCas9 in PGM1 medium on Matrigel-coated plates.
- Pre-treat with doxycycline (1-2 μg/mL) 24 hours before nucleofection to induce Cas9 expression.
Nucleofection
- Dissociate cells with EDTA to maintain viability.
- Combine 5μg modified sgRNA with 8×10⁵ cells in P3 nucleofection buffer.
- Electroporate using Lonza 4D-Nucleofector with program CA-137.
- Plate recovered cells in pre-warmed medium with ROCK inhibitor.
Efficiency Analysis
- Harvest cells 72-96 hours post-nucleofection for initial efficiency assessment.
- For low efficiency targets (<50%), repeat nucleofection following same procedure.
- Analyze INDELs using ICE algorithm with Sanger sequencing data.
Validation:
- This optimized system achieved over 80% efficiency for double-gene knockouts and up to 37.5% homozygous knockout efficiency for large fragment deletions [12].
- Western blot integration enables rapid detection of ineffective sgRNAs that produce high INDEL rates but retain protein expression.
Pathway Manipulation for Enhanced Knockout Efficiency
Strategic manipulation of DNA repair pathways can bias outcomes toward desired mutational profiles. Research demonstrates that chemical or genetic perturbations can direct DNA repair toward desired editing outcomes across diverse cell types including neurons, cardiomyocytes, and primary T cells [54].
Small Molecule Interventions:
- DNA-PK inhibitors: Enhance cell death in aberrant cells when combined with CRISPR targeting, particularly effective in cancer cell lines [77].
- ATM/ATR inhibitors: Sensitize cells to DNA damage and can alter repair pathway choice.
- PARP inhibitors: Exploit synthetic lethality in HR-deficient backgrounds and can modulate repair dynamics.
Critical Consideration: The effect of pathway manipulation is highly cell type-specific. The same intervention may produce opposite effects in different cellular contexts, necessitating empirical optimization for each model system.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Optimized CRISPR Knockout Generation
| Reagent Category | Specific Examples | Function & Application | Cell Type Specificity |
|---|---|---|---|
| CRISPR Delivery Systems | Lentiviral vectors (LentiCRISPRv2), Virus-like particles (VLPs), Lipofectamine 3000 | Efficient RNP delivery with varying persistence | Lentivirus: hard-to-transfect cells; VLPs: postmitotic cells; Lipofection: standard cell lines |
| Selection Agents | Puromycin, Blasticidin, GFP/RFP sorters | Enrichment of successfully transfected cells | Concentration must be optimized per cell line via kill curve assays |
| Repair Pathway Modulators | DNA-PK inhibitors (NU7441), ATM inhibitors (KU-60019), PARP inhibitors (Olaparib) | Bias DNA repair toward specific pathways to control editing outcomes | HR-deficient cells show particular sensitivity to PARP inhibitors |
| Validation Tools | T7E1 assay, TIDE analysis, ICE analysis, Western blotting | Confirm editing efficiency and protein-level knockout | Western blot essential for detecting ineffective sgRNAs |
| Cell Culture Supplements | ROCK inhibitor (Y-27632), Lenti-X concentrator, Polybrene | Enhance cell survival post-transfection and improve viral transduction | ROCK inhibitor critical for sensitive primary and stem cells |
DNA Repair Pathway Signaling Schematic
DNA Repair Pathway Decisions After CRISPR Cutting
Experimental Workflow for Cell Line-Specific Optimization
Optimized Workflow for CRISPR Knockout Generation
Successful CRISPR knockout generation requires moving beyond one-size-fits-all protocols to embrace cell line-specific optimization strategies. The interplay between cellular identity, DNA repair machinery, and experimental parameters dictates editing outcomes and clonal survival. By applying the principles and protocols outlined in this application note—including appropriate delivery methods, optimized timelines reflecting cell-specific repair kinetics, and strategic pathway manipulation—researchers can significantly improve the efficiency and reliability of knockout cell line generation across diverse experimental systems.
Ensuring Fidelity: Comprehensive Validation of Knockout Cell Lines and Functional Assays
In the field of CRISPR-Cas9-mediated knockout cell line generation, robust genomic validation is paramount for confirming intended genetic modifications and detecting potential unintended alterations. The journey from Sanger sequencing to targeted Next-Generation Sequencing (NGS) represents a significant evolution in verification methodologies, each offering distinct advantages for researchers characterizing engineered cell models. As CRISPR-Cas9 technology revolutionizes biological research and drug discovery, the choice of validation strategy directly impacts the reliability, depth, and safety assessment of resulting cell lines [8] [73]. This application note delineates structured protocols and comparative analyses to guide researchers in selecting and implementing appropriate validation strategies within the context of CRISPR-Cas9 research, with particular emphasis on applications relevant to drug development professionals.
The fundamental goal of genomic validation in CRISPR workflows is to confirm the presence of intended insertions or deletions (indels) while screening for off-target effects that could compromise experimental results or therapeutic safety [73] [78]. While Sanger sequencing provides a cost-effective method for initial confirmation of editing at specific loci, the emergence of targeted NGS enables comprehensive assessment of editing efficiency, heterogeneity, and potential structural variations across multiple genomic regions simultaneously [79] [80]. The integration of these complementary approaches provides researchers with a multi-tiered validation framework that balances throughput, sensitivity, and cost-efficiency.
Comparative Analysis of Sequencing Methodologies
Technical Specifications and Performance Metrics
The selection between Sanger sequencing and targeted NGS requires careful consideration of performance specifications relative to experimental objectives. The table below summarizes the key technical parameters of each method:
Table 1: Performance Comparison of Sanger Sequencing and Targeted NGS
| Parameter | Sanger Sequencing | Targeted NGS |
|---|---|---|
| Fundamental Method | Chain termination using ddNTPs [81] | Massively parallel sequencing [81] |
| Maximum Throughput | Single fragment per reaction [82] | Millions to billions of reads per run [81] [80] |
| Read Length | 500-1000 bp [81] [82] | 50-300 bp (short-read); >10,000 bp (long-read) [81] [80] |
| Variant Detection Sensitivity | 15-20% [83] | 1-5% [83] |
| Error Rate | ~0.001% [83] | 0.1-15% (platform-dependent) [80] [83] |
| Best Applications | Single-gene validation, confirmation of known variants [81] [82] | Detection of complex indels, off-target effects, structural variations [79] [73] |
Economic and Operational Considerations
The economic evaluation of sequencing methodologies extends beyond per-sample costs to encompass infrastructure requirements, personnel expertise, and project scalability. Sanger sequencing maintains advantages for low-throughput applications with minimal bioinformatics overhead, while targeted NGS achieves superior cost-efficiency at scale despite higher initial investment [81] [82].
Table 2: Economic and Operational Considerations
| Factor | Sanger Sequencing | Targeted NGS |
|---|---|---|
| Cost Profile | Low capital investment, high cost per base for large projects [81] [82] | High capital investment, low cost per base for large projects [81] [82] |
| Equipment Cost | $10,000-$50,000 | $50,000-$1,000,000+ [82] |
| Bioinformatics Requirements | Minimal (basic sequence alignment) [81] | Extensive (specialized pipelines for alignment, variant calling) [81] [80] |
| Personnel Expertise | Standard molecular biology techniques | Advanced computational and statistical skills [82] |
| Optimal Project Scale | 1-10 targets [82] | 10+ targets or genome-wide analyses [81] |
Integrated Validation Workflow for CRISPR-Cas9 Knockouts
The following workflow diagram illustrates the integrated validation approach for CRISPR-Cas9 knockout cell lines, combining both Sanger sequencing and targeted NGS methodologies at appropriate stages:
Detailed Experimental Protocols
Protocol 1: Sanger Sequencing Validation with Computational Analysis
This protocol details the procedure for validating CRISPR-Cas9 editing efficiency using Sanger sequencing followed by computational decomposition of sequencing traces [79].
Materials and Reagents:
- PCR reagents (primers, polymerase, dNTPs)
- Gel extraction kit
- Sanger sequencing reagents or commercial service
- TIDE, ICE, or DECODR web tool access [79]
Procedure:
- Design PCR primers flanking the CRISPR target site (amplicon size: 300-500 bp)
- Amplify target region from wild-type and edited cell populations
- Purify PCR products using gel extraction kit
- Submit samples for Sanger sequencing with appropriate primers
- Upload sequencing data to computational tool:
- TIDE (Tracking of Indels by Decomposition)
- ICE (Inference of CRISPR Edits)
- DECODR (Deconvolution of Complex DNA Repair) [79]
- Analyze decomposition results for indel spectrum and efficiency
Technical Notes:
- Computational tools show highest accuracy with simple indels of few base changes [79]
- DECODR demonstrates superior performance for identifying specific indel sequences [79]
- For complex edits or knock-ins, TIDER (TIDE for Knock-Ins) provides enhanced accuracy [79]
Protocol 2: Targeted NGS for Comprehensive Off-Target Assessment
This protocol describes a targeted NGS approach to identify off-target effects and structural variations in CRISPR-edited cell lines [73].
Materials and Reagents:
- DNA extraction kit
- Targeted NGS panel (including on-target sites and predicted off-targets)
- Library preparation reagents
- NGS platform (Illumina, PacBio, or Oxford Nanopore)
Procedure:
- Extract high-quality genomic DNA from edited and control cells
- Design capture panel encompassing:
- Primary target site(s)
- Top 50-100 predicted off-target sites (using tools like Cas-OFFinder)
- Regions with high sequence similarity to target
- Flanking sequences (extend ≥1 kb from cut site) [73]
- Prepare sequencing libraries using manufacturer's protocol
- Sequence with minimum coverage of 100X for variant detection
- Analyze data with specialized bioinformatics pipeline:
- Map reads to reference genome
- Call variants in target regions
- Identify structural variations using SV detection algorithms
- Compare with control sample to filter background mutations [73]
Technical Notes:
- Include positive and negative controls in each sequencing run
- For structural variation detection, ensure sufficient read length (>150 bp) or use long-read technologies [73]
- Pay particular attention to kilobase- to megabase-scale deletions that may eliminate primer binding sites and evade detection in standard PCR-based assays [73]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Genomic Validation
| Reagent/Category | Specific Examples | Function in Workflow |
|---|---|---|
| CRISPR Delivery Vectors | pX459 [8], LentiCRISPRv2 [7] | Delivery of Cas9 and guide RNA to target cells |
| Selection Agents | Puromycin [8] [7] | Selection of successfully transfected cells |
| Computational Design Tools | Synthego CRISPR Design Tool [7], CRISPOR | Guide RNA design and off-target prediction |
| Sanger Analysis Tools | TIDE, ICE, DECODR [79] | Decomposition of sequencing traces to quantify editing |
| Targeted NGS Panels | Custom hybrid capture panels | Enrichment of genomic regions of interest prior to sequencing |
| Variant Callers | GATK, FreeBayes | Identification of genetic variants from NGS data |
| Structural Variation Detectors | Manta, DELLY | Detection of large-scale genomic rearrangements |
Advanced Considerations for Drug Development Applications
Safety Assessment and Structural Variation Detection
Beyond confirming intended on-target edits, comprehensive genomic validation must address the risk of structural variations (SVs) that pose significant safety concerns for therapeutic development [73]. Recent studies reveal that CRISPR-Cas9 can induce kilobase- to megabase-scale deletions, chromosomal translocations, and complex rearrangements at frequencies higher than previously appreciated [73].
The following diagram illustrates the spectrum of genetic outcomes that must be considered in safety assessment:
Detection of these aberrations requires specialized approaches, as traditional short-read amplicon sequencing may miss large deletions that eliminate primer binding sites [73]. Recommended strategies include:
- Long-range PCR followed by NGS to detect intermediate-sized deletions
- Whole-genome sequencing with long-read technologies (Oxford Nanopore, PacBio) for comprehensive SV assessment [80]
- CAST-Seq or LAM-HTGTS for sensitive detection of chromosomal translocations [73]
Mitigation Strategies for High-Risk Applications
For preclinical and therapeutic applications, implement additional risk mitigation strategies:
- Avoid DNA-PKcs inhibitors (e.g., AZD7648) that exacerbate genomic aberrations [73]
- Utilize high-fidelity Cas9 variants (e.g., HiFi Cas9) to reduce off-target effects [73]
- Employ computational prediction tools during gRNA design to avoid sites with potential for structural variations
- Conduct rigorous clone screening with multiple validation methods before selection for expansion
The evolution of genomic validation from Sanger sequencing to targeted NGS provides researchers with a powerful toolkit for comprehensive characterization of CRISPR-Cas9 edited cell lines. While Sanger sequencing remains the gold standard for initial confirmation of editing efficiency at specific loci, targeted NGS enables deeper analysis of editing heterogeneity, off-target effects, and structural variations critical for safety assessment. An integrated approach that leverages both methodologies at appropriate stages of the validation workflow offers the most robust strategy for generating high-quality knockout cell lines suitable for research and drug development applications. As CRISPR technologies continue to advance toward clinical applications, comprehensive genomic validation will play an increasingly vital role in ensuring the efficacy and safety of genetically engineered cell models.
Within the broader context of CRISPR-Cas9 knockout (KO) cell line generation research, confirming successful protein-level knockdown represents a critical validation checkpoint. The generation of a complete KO cell line is a complex process that requires significant time investment, often taking nearly five months, with most researchers needing to restart experiments multiple times before achieving their desired knock-out cell lines [14]. After navigating challenges in guide RNA design, transfection, and clonal isolation, rigorous confirmation of the intended genetic edit at the protein level becomes paramount for ensuring downstream experimental validity.
This application note details two principal methodologies—Western blot (immunoblotting) and mass spectrometry (MS)-based proteomics—for the verification of protein knockdown in CRISPR-engineered cell lines. We provide detailed protocols, strategic implementation guidelines, and resource information to equip researchers with the tools necessary for robust proteomic confirmation of their gene edits.
Western Blot for Knockdown Verification
Western blotting remains a widely accessible and fundamental technique for detecting the presence or absence of a target protein. Its proper application, however, requires careful optimization and validation to generate reliable, quantitative data.
Critical Pre-requisite: Antibody Validation
A primary source of irreproducible research findings stems from the use of poorly characterized antibody reagents [84]. Antibody validation is the process of confirming that an antibody recognizes your protein of interest with low cross-reactivity to other targets, and it is critical for ensuring consistent, reproducible Western blot results [85]. The International Working Group for Antibody Validation (IWGAV) recommends several strategies, with genetic validation being the gold standard for Western blotting [84].
Key Antibody Validation Strategies:
- Genetic Strategies (Gold Standard): This involves measuring the signal in control cells versus cells in which the target protein has been knocked out (e.g., via CRISPR-Cas9) or knocked down (e.g., via RNAi). In a valid KO cell lysate, there should be very little to no signal, and any signal observed indicates cross-reactivity or non-specific binding [85] [84].
- Independent Antibody Strategies: Using two or more different antibodies that recognize independent epitopes on the same target protein can gauge specificity. Correlation between the results increases confidence that the observed signal is specific to the target [85] [84].
- Orthogonal Strategies: Comparing the quantification from Western blot with an antibody-independent method (e.g., targeted mass spectrometry) for several samples can validate the antibody's performance [85].
- Expression of Tagged Proteins: Expressing the target protein with an affinity tag (e.g., FLAG, GFP) and matching the antibody signal to the tag's signal can serve as a validation method, though overexpression may artificially mask off-target binding [85].
Detailed Western Blot Protocol for Knockdown Confirmation
The following protocol is systematized to minimize variability and enhance quantitability [86].
Step-by-Step Workflow:
Protein Extraction and Quantification:
- Lyse control (wild-type) and putative KO clonal cell lines using an appropriate lysis buffer. Include a protease inhibitor cocktail.
- Quantify protein concentration using a compatible assay (e.g., Bradford assay). Critical: Use the same total protein amount for all samples to be compared. Normalization at this stage is crucial for later quantitative analysis.
Gel Electrophoresis and Transfer:
- Separate proteins by SDS-PAGE. Include a pre-stained protein ladder.
- Transfer proteins to a nitrocellulose or PVDF membrane. Verify efficient transfer using reversible stains like Ponceau S.
Immunoblotting:
- Block the membrane with 5% non-fat dry milk or BSA in TBST for 1 hour at room temperature.
- Incubate with the validated primary antibody, diluted in blocking buffer, overnight at 4°C with gentle agitation.
- Wash the membrane 3 times for 5 minutes each with TBST.
- Incubate with an appropriate HRP-conjugated or fluorescently-labeled secondary antibody for 1 hour at room temperature.
- Perform a final series of 3 washes with TBST.
Detection and Analysis:
- Develop the blot using chemiluminescent or fluorescent substrates and image with a digital imager.
- For quantitative Western blotting, ensure the signal is within the linear range of detection for both the target and loading control [86].
- Critical Controls:
- Positive Control: Lysate from a cell line known to express the target protein.
- Negative Control: Your validated KO cell line lysate. This is essential for confirming the absence of the target and the specificity of the antibody.
- Loading Control: Probe for a constitutively expressed housekeeping protein (e.g., β-Actin, Tubulin, GAPDH) to ensure equal loading.
The diagram below illustrates the logical workflow and critical control points for using Western blot to verify protein knockdown.
Mass Spectrometry for Knockdown Verification
Mass spectrometry offers an antibody-independent, highly specific method for confirming protein knockdown. It can provide absolute or relative quantification and is particularly powerful for verifying the complete absence of a protein.
Mass Spectrometry-Based Quantification Workflow
A highly specific approach involves combining Multiple Reaction Monitoring (MRM) with Protein-AQUA (Absolute QUAntification) using isotopically labeled synthetic peptides as internal standards [87].
Detailed MRM / Protein-AQUA Protocol:
Sample Preparation:
- Harvest control and KO cells. Lyse cells and digest chromosomal DNA with Benzonase Nuclease.
- Determine the protein concentration of the lysate using a Bradford assay [87].
Protein Digestion and Spike-In of Standards:
- Reduce, alkylate, and digest proteins (e.g., with trypsin) from a fixed amount of lysate (e.g., 30 μg).
- Spike-in AQUA Peptides: Synthesize and purify heavy isotopically labeled (>98% 13C, 15N) versions of target peptides specific to your protein of interest. Spike a known amount (e.g., 5 pmol) of these AQUA peptides into the digested protein sample [87].
LC-MRM Analysis:
- Introduce the sample into a triple quadrupole mass spectrometer via Liquid Chromatography (LC).
- Q1 (Quadrupole 1): Select the precursor ion of a specific peptide (both endogenous and heavy AQUA).
- q2 (Collision Cell): Fragment the selected precursor ion.
- Q3 (Quadrupole 3): Monitor a specific, predictable fragment ion from the peptide.
- This process, defined for a precursor ion -> fragment ion pair, is a Transition. Monitoring several transitions in parallel constitutes MRM [87].
Quantification and Data Analysis:
- The chromatographic peak areas for the endogenous light peptide and the spiked heavy AQUA peptide are integrated.
- The ratio of the endogenous peptide peak area to the AQUA peptide peak area is used to calculate the absolute amount of the target protein in the original sample.
- A successful knockdown is indicated by a drastic reduction or absence of the endogenous peptide signal in the KO sample compared to the control, while the AQUA peptide signal remains constant.
The workflow for this targeted mass spectrometry approach is outlined below.
Comparative Analysis of Techniques
The choice between Western blot and mass spectrometry depends on the experimental requirements, available resources, and the required level of specificity and quantification.
Table 1: Comparison of Western Blot and Mass Spectrometry for Knockdown Verification
| Feature | Western Blot | Mass Spectrometry (MRM/AQUA) |
|---|---|---|
| Principle | Antibody-based detection of proteins [14] [88] | Detection and quantification of proteolytic peptides via mass/charge [87] [88] |
| Specificity | Dependent on antibody specificity; requires rigorous validation [85] [84] | Very high; based on precursor ion mass and a unique fragment ion [87] |
| Quantitation | Semi-quantitative; requires careful controls and linear range detection [86] | Highly quantitative; enables absolute quantification with isotope standards [87] |
| Throughput | Relatively high; can multiplex several samples on one gel | Moderate; requires individual LC-MS runs per sample |
| Key Advantage | Accessibility, cost-effectiveness, visual confirmation of protein size | Antibody-independent, highly specific, can distinguish isoforms/PTMs |
| Key Limitation | Susceptible to antibody cross-reactivity and lot-to-lot variability [84] | Higher cost, requires specialized instrumentation and expertise |
| Ideal Use Case | Initial, rapid screening of multiple clones | Definitive, gold-standard validation of complete knockout |
The Scientist's Toolkit: Research Reagent Solutions
Successful verification relies on high-quality reagents and tools. The following table summarizes essential materials and their functions.
Table 2: Essential Research Reagents and Resources for Knockdown Verification
| Category | Reagent / Resource | Function and Importance |
|---|---|---|
| CRISPR Validation | KO cell line lysate | Critical negative control for validating antibody specificity in Western blot [85] [84] |
| Antibody Resources | Validated primary antibodies | Ensure specificity; seek vendors that provide application-specific validation data [84] |
| Recombinant antibodies | Recommended to minimize batch-to-batch variation [84] | |
| Mass Spectrometry | AQUA Peptides | Isotopically labeled internal standards for absolute protein quantification by MS [87] |
| Triple Quadrupole Mass Spectrometer | Instrument platform for highly sensitive and specific MRM assays [87] | |
| Informatic Resources | CRISPOR tool | For designing specific sgRNAs during the CRISPR cell line generation phase [61] |
| Expression Atlas / Human Protein Atlas | To check expected protein expression patterns in control cell lines [84] |
Within the multi-step workflow of CRISPR-Cas9 KO cell line generation, proteomic confirmation of the knockout is a non-negotiable step that validates the entire preceding effort. While Western blotting serves as a powerful and accessible tool for initial screening, its reliability is entirely contingent upon the use of rigorously validated antibodies. Mass spectrometry, particularly targeted MRM with isotopic standards, provides an orthogonal, antibody-independent method for definitive verification and absolute quantification.
A combined approach, utilizing Western blot for initial clonal screening followed by mass spectrometry for definitive confirmation of lead clones, represents the most robust strategy. This ensures that subsequent functional studies in drug development and basic research are built upon a foundation of genetically and proteomically verified cellular models. Adhering to the detailed protocols and validation strategies outlined here will significantly enhance the reproducibility and reliability of research findings.
Functional phenotyping represents a critical bridge between genetic manipulation and the understanding of disease biology. In the context of CRISPR-Cas9 knockout (CRISPRko) cell line generation, it provides the methodological framework for assessing how genetic perturbations manifest as observable cellular characteristics. As pharmaceutical and biotechnology companies increasingly rely on precise gene-modified cell lines for target validation and drug discovery, robust functional phenotyping protocols have become indispensable for translating genetic edits into biologically and therapeutically meaningful insights [21].
This application note details established and emerging methodologies for comprehensive functional assessment of CRISPR-generated cell models, providing researchers with standardized protocols for evaluating the biological consequences of genetic manipulations in disease-relevant contexts.
Methodological Approaches for Functional Phenotyping
Single-Cell Multi-Omic Phenotyping
Single-cell DNA–RNA sequencing (SDR-seq) represents a significant advancement for simultaneously genotyping cells and assessing transcriptomic consequences of genetic variants. This method enables accurate determination of variant zygosity alongside associated gene expression changes at single-cell resolution [89] [90].
Experimental Protocol: SDR-seq for Concurrent Genotype-Phenotype Analysis
- Cell Preparation: Dissociate cells into a single-cell suspension. Fix and permeabilize cells using glyoxal (which provides superior RNA quality compared to PFA by avoiding nucleic acid cross-linking) [89].
- In Situ Reverse Transcription: Perform reverse transcription using custom poly(dT) primers containing unique molecular identifiers (UMIs), sample barcodes (BCs), and capture sequences (CSs) to label cDNA molecules [89].
- Droplet-Based Partitioning: Load cells onto a microfluidic system (e.g., Tapestri from Mission Bio). Generate first droplets, then lyse cells and treat with proteinase K. Mix with reverse primers for intended gDNA and RNA targets [89].
- Multiplexed PCR Amplification: During second droplet generation, introduce forward primers with CS overhangs, PCR reagents, and barcoding beads containing cell BC oligonucleotides. Perform multiplexed PCR to simultaneously amplify gDNA and RNA targets within each droplet [89].
- Library Preparation and Sequencing: Separate gDNA and RNA libraries using distinct overhangs on reverse primers. Sequence gDNA libraries for full-length variant coverage and RNA libraries for transcript expression information with cell BC, sample BC, and UMI preservation [89].
Table 1: SDR-seq Performance Metrics Across Panel Sizes
| Panel Size (Targets) | gDNA Target Detection Rate | RNA Target Detection Rate | Cells Analyzed per Run |
|---|---|---|---|
| 120 (60 gDNA/60 RNA) | >80% | >80% | Thousands |
| 240 (120 gDNA/120 RNA) | >80% | >80% | Thousands |
| 480 (240 gDNA/240 RNA) | ~80% (minor decrease) | ~80% (minor decrease) | Thousands |
Image-Based Morphological and Dynamic Phenotyping
CellPhe provides a pattern recognition toolkit for unbiased characterization of cellular phenotypes from time-lapse imaging data, enabling quantification of morphology, texture, and dynamics in CRISPR-modified cells [91].
Experimental Protocol: CellPhe for Longitudinal Phenotypic Analysis
- Image Acquisition and Preprocessing: Capture time-lapse videos using preferred imaging modality (e.g., fluorescence, ptychography). Process images through segmentation and tracking algorithms to identify individual cells and follow them over time. Export cell boundary coordinates for analysis [91].
- Feature Extraction: Calculate an extensive list of morphology features (size, shape), texture features, and dynamical features (movement, division patterns) from the cell boundary data across all timepoints [91].
- Segmentation Error Removal: Implement automated classification to identify and remove erroneous cell boundaries resulting from inaccurate tracking or segmentation. This quality control step is crucial for reliable downstream analysis [91].
- Feature Selection and Analysis: Apply customized feature selection to identify variables providing greatest discrimination between experimental conditions. Use ensemble classification for phenotype prediction and clustering algorithms for identifying heterogeneous cellular subpopulations [91].
Table 2: CellPhe Segmentation Error Removal Performance
| Cell Line | Training Set (Errors/Total) | Test Set Errors Identified | Accuracy | Common Error Types |
|---|---|---|---|---|
| MDA-MB-231 | 241/1,942 | 223/1,478 | 97.3% | Under/over-segmentation, background pickup, ID swapping |
| MCF-7 | 192/500 | 188/692 | >95% | Mitosis tracking errors, size estimation failures |
Pooled CRISPR Screening with Functional Readouts
Perturbomics combines pooled CRISPR screening with high-content phenotypic readouts to systematically link genetic perturbations to functional consequences at scale [92].
Experimental Protocol: Pooled CRISPRko Screening for Essential Gene Identification
- gRNA Library Design and Delivery: Design a genome-wide or focused gRNA library targeting genes of interest. Clone the library into lentiviral vectors and transduce at low MOI (<0.3) into Cas9-expressing cells to ensure single gRNA integration per cell [93] [92].
- Selection Pressure Application: Subject the pooled cell population to selective pressures relevant to the biological question (e.g., drug treatment, nutrient deprivation, FACS sorting based on surface markers) [92].
- Genomic DNA Extraction and Sequencing: After selection, extract genomic DNA from surviving cell populations. Amplify and sequence integrated gRNA cassettes to determine relative abundance changes [93].
- Bioinformatic Analysis: Process sequencing data using specialized computational tools (e.g., MAGeCK, BAGEL) to identify significantly enriched or depleted gRNAs, indicating genes essential for survival under the selection pressure [93].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Platforms for Functional Phenotyping
| Reagent/Platform | Primary Function | Application Context |
|---|---|---|
| CRISPRko Systems (SpCas9, AsCas12a) | Indels via NHEJ; precise editing via HDR | Generation of loss-of-function models; target validation [94] |
| CRISPRi/a Systems (dCas9-KRAB, dCas9-VPR) | Gene knockdown/activation without DSBs | Essential gene screening; functional analysis of lncRNAs [92] |
| Base/Prime Editors (ABE, CBE, PE) | Precise nucleotide conversion without DSBs | Modeling point mutations; functional analysis of SNVs [94] |
| Tapestri Platform (Mission Bio) | Single-cell DNA+RNA sequencing | Genotype-phenotype linkage at single-cell resolution [89] |
| CellPhe Toolkit | Pattern recognition from time-lapse imaging | Morphological and dynamic phenotyping [91] |
| MAGeCK Computational Pipeline | Bioinformatics analysis of CRISPR screens | Hit identification from pooled screens [93] |
Discussion and Future Perspectives
Functional phenotyping technologies are evolving toward increasingly multiplexed, high-resolution, and dynamic assessments of cellular responses. The integration of artificial intelligence and machine learning with functional phenotyping is already enhancing predictive modeling for gRNA design and automating analysis of complex cellular phenotypes [21]. Emerging methods like single-cell multi-omics and high-content live-cell imaging are pushing the boundaries of what can be measured from individual CRISPR-modified cells.
Each phenotyping approach offers complementary strengths. Pooled CRISPR screens provide scalability for genome-wide investigations, image-based methods capture spatial and temporal dynamics, and multi-omic approaches enable deep molecular characterization. The strategic selection and integration of these methodologies will accelerate the translation of CRISPR-generated disease models into biologically meaningful insights and therapeutic breakthroughs.
As these technologies mature, they are transforming functional phenotyping from a descriptive exercise to a predictive science, ultimately enhancing our ability to connect genetic alterations to phenotypic outcomes in disease models and drug development pipelines.
The development of robust potency assays is a critical regulatory requirement for the clinical advancement and commercialization of recombinant adeno-associated virus (rAAV) gene therapy products. Potency assays quantitatively measure the biological activity of a drug product that is linked to its intended mechanism of action (MoA) and clinical efficacy [95]. For rAAV-based therapies targeting monogenic disorders, a key challenge has been the development of cell-based potency assays that accurately reflect the biological function of the transgene in a physiologically relevant context. This case study details the development of a novel cell-based potency assay for an rAAV gene therapy product targeting Limb-Girdle Muscular Dystrophy R9 (LGMDR9), a progressive neuromuscular disorder caused by mutations in the Fukutin-related protein (FKRP) gene [96].
LGMDR9 is characterized by deficient glycosylation of alpha-dystroglycan (α-DG), a key membrane protein essential for maintaining muscle integrity [96]. The biological MoA of the rAAV-FKRP product is the restoration of functional FKRP protein, which subsequently enables proper α-DG glycosylation. Traditional potency testing approaches using patient-derived cells faced limitations due to residual α-DG glycosylation present in these cells, which obscured a clear assessment of glycosylation restoration following rAAV-FKRP treatment [96]. To address this challenge, we employed CRISPR-Cas9 technology to generate a completely novel FKRP knockout (KO) muscle cell line fully depleted of α-DG glycosylation, thereby creating a optimized cellular system for quantifying the biological activity of rAAV-FKRP products through measurement of α-DG glycosylation restoration.
Material and Methods
Research Reagent Solutions
Table 1: Essential Research Reagents for Knockout Cell Line Generation and Potency Assays
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Cell Lines | Human immortalized myoblasts (AB1190) [96], HEK293 [97] | Provide cellular substrate for gene editing and potency assessment; selected for relevance to MoA and transduction efficiency. |
| CRISPR-Cas9 System | pSpCas9(BB)-2A-GFP (PX458) [98], sgRNAs targeting FKRP translation initiation site [96] | Enables precise genome editing through induction of double-strand breaks at specific genomic loci. |
| Cell Culture | Skeletal Muscle Cell Growth/Differentiation Media [96], Collagen I-coated plates [96] | Supports propagation and maintenance of muscle cells and promotes differentiation into myotubes. |
| Transfection Reagent | Lipofectamine RNAi MAX [98] | Facilitates delivery of CRISPR-Cas9 components into mammalian cells. |
| Critical Assay Reagents | Primary antibodies against glycosylated α-DG [96], IRDye fluorescent secondary antibodies [96], LC-MS/MS systems [97] | Enable detection and quantification of the key biological response (glycosylation restoration). |
| Reference Standard | Well-characterized rAAV-FKRP batch [96] [95] | Serves as comparator for test articles in relative potency determination; essential for assay calibration. |
Generation of FKRP Knockout Cell Line Using CRISPR-Cas9
The generation of a clonal FKRP knockout muscle cell line involved a multi-step process to ensure complete loss of gene function and subsequent validation.
2.2.1 sgRNA Design and Vector Construction. A single-guide RNA (sgRNA) was designed to target the translation initiation site of the human FKRP gene to ensure complete disruption of protein function [96]. The sgRNA protospacer sequence was cloned into the pSpCas9(BB)-2A-GFP (PX458) plasmid, which co-expresses the sgRNA, Streptococcus pyogenes Cas9 nuclease, and a GFP reporter gene enabling fluorescence-based enrichment of transfected cells [98].
2.2.2 Cell Transfection and Clonal Isolation. Human immortalized myoblasts (AB1190 wild-type line) were cultured in Skeletal Muscle Cell Growth Medium on collagen I-coated plates. At approximately 70% confluence, cells were transfected with the CRISPR-Cas9 plasmid construct using a lipid-based nanoparticle delivery system [96] [98]. Forty-eight hours post-transfection, GFP-positive cells were isolated using fluorescence-activated cell sorting (FACS) and deposited as single cells into 96-well plates for clonal expansion [99]. Clones were maintained under standard culture conditions (37°C, 5% CO2) with periodic medium changes until sufficient cell numbers were achieved for genotypic and phenotypic characterization.
2.2.3 Genotypic Validation of Knockout Clones. Genomic DNA was isolated from expanded clonal populations. The target region of the FKRP gene was amplified by PCR and analyzed using next-generation sequencing (NGS) to identify insertion/deletion (indel) mutations. Clones exhibiting frameshift mutations in both FKRP alleles were selected as candidate knockout lines [96] [98]. Sanger sequencing, analyzed with tools such as ICE (Inference of CRISPR Edits), can provide initial screening but may lack sensitivity for detecting longer indels and accurately determining allele frequencies in a mixed population [98].
2.2.4 Phenotypic Validation. Selected clones were differentiated into myotubes over four days using differentiation-specific medium. The successful knockout of FKRP and the resulting functional deficit were confirmed by demonstrating a complete absence of α-DG glycosylation via Western blot analysis using antibodies specific to glycosylated α-DG [96]. This phenotypic confirmation is crucial, as it verifies that the genetic editing resulted in the intended functional outcome, establishing a clean background for the potency assay.
Development of the On-Cell Western Potency Assay
The cell-based potency assay was developed to measure the biological activity of rAAV-FKRP products by quantifying the restoration of α-DG glycosylation in the validated KO-FKRP cell line.
2.3.1 Cell Seeding and Differentiation. KO-FKRP myoblasts were seeded at a density of 9.6 × 10^4 cells per well in 96-well plates pre-coated with collagen I. After 48 hours, the growth medium was replaced with differentiation medium to induce myotube formation over four days [96].
2.3.2 rAAV-FKRP Transduction. Differentiated myotubes were transduced with serial dilutions of the rAAV-FKRP test article and a reference standard. The assay utilized a range of multiplicities of infection (MOIs) to generate a dose-response curve [96]. A similar approach for an AAV2-hRPE65v2 potency assay used nine MOI levels to fit a 3-parameter logistic model [97].
2.3.3 On-Cell Western Detection. Following an appropriate incubation period to allow for transgene expression and functional activity, cells were fixed and immunostained. The high-throughput On-Cell Western methodology was employed, which involves incubating the fixed cells with a primary antibody against fully glycosylated α-DG, followed by an IRDye-labeled secondary antibody. The signal was quantified using an infrared imaging system [96]. This method combines the specificity of Western blotting with the throughput capabilities of an ELISA, making it suitable for a potency assay requiring reproducibility and scalability.
2.3.4 Data Analysis and Relative Potency Calculation. The fluorescence signal for each dilution was plotted against the log10-transformed vector concentration to generate a dose-response curve. The half maximal effective concentration (EC50) was determined by fitting the data to a 4- or 5-parameter logistic (4-PL/5-PL) model [95]. The relative potency of the test article was calculated by comparing its EC50 to that of the reference standard using parallel-line analysis [96] [97].
Diagram 1: Experimental workflow for developing and implementing the rAAV-FKRP potency assay, from knockout cell line generation to final potency determination.
Results and Data Analysis
Characterization of the FKRP Knockout Cell Line
Genotypic analysis by NGS confirmed the presence of frameshift indels in both FKRP alleles in the selected clones, resulting in premature termination codons and predicted loss of protein function. Phenotypic validation via Western blot demonstrated a complete absence of glycosylated α-DG in the knockout clones, while wild-type control cells showed a strong signal. This confirmed the creation of a cellular model fully depleted of the target glycoform, providing a stringent system for quantifying functional FKRP activity [96].
On-Cell Western Potency Assay Performance
The developed On-Cell Western assay successfully detected a dose-dependent increase in glycosylated α-DG signal in response to increasing concentrations of rAAV-FKRP. The dose-response curves exhibited a sigmoidal shape, allowing for reliable EC50 calculation. The determination of the EC50 provided a robust method for comparing the biological activity of different rAAV-FKRP batches against a reference standard [96]. The use of a KO cell line eliminated the background signal from residual glycosylation, thereby increasing the assay's dynamic range and sensitivity compared to assays using patient-derived cells.
Table 2: Key Validation Parameters for Cell-Based Potency Assays (Based on Regulatory Guidance and Case Studies)
| Validation Parameter | Description | Exemplary Acceptance Criteria [97] |
|---|---|---|
| System and Sample Suitability | Ensures valid and reliable assay performance in each run. | 3PL model fit with multiple doses; 90% CI for relative potency within 76%-130%. |
| Specificity | Ability to distinguish the active product from inactive components. | Formulation buffer shows no dose-response; test article and reference show parallel dose-response. |
| Precision | Degree of reproducibility of measured potency under defined conditions. | %GCV for intermediate precision <30% at each potency level. |
| Relative Accuracy | Closeness of the measured relative potency to the expected value. | Relative bias within ±15% at all tested levels. |
| Linearity & Range | Ability to produce results proportional to analyte concentration within a given range. | Demonstrated from 50% to 150% of nominal concentration; R² ≥ 0.85. |
| Robustness | Capacity to remain unaffected by small, deliberate variations in procedural parameters. | Assay remains valid under specified variations (e.g., transduction time ± 4h). |
Assay Validation Strategy
To be suitable for use as a lot-release test for a commercial gene therapy product, the potency assay must undergo formal validation. The validation of the AAV2-hRPE65v2 potency assay, which provides a regulatory benchmark, adhered to USP <1033> guidelines and assessed the parameters listed in Table 2 [97]. The validation involved testing multiple relative potency levels (e.g., 50%, 75%, 100%, 125%, 150%) to ensure accuracy across the reportable range. The results demonstrated that the assay consistently met pre-defined acceptance criteria, ensuring its precision, accuracy, and reproducibility for regulatory compliance [97].
Diagram 2: Signaling pathway of FKRP mutation and mechanism of action of rAAV-FKRP gene therapy. The disease pathology (red) stems from FKRP defects leading to loss of α-DG function. The therapy (green) restores the functional glycosylation of α-DG, which is the direct readout of the potency assay.
Discussion
The integration of CRISPR-Cas9-generated knockout cell lines into the development of cell-based potency assays represents a significant advancement for the gene therapy field. This approach directly addresses a major challenge in potency testing for recessive genetic disorders: the presence of residual protein function or background signal in patient-derived cells, which can obscure a clear and quantitative measurement of the therapeutic product's biological activity [96]. By creating a null background, the assay's dynamic range and sensitivity are maximized, allowing for precise quantification of the transgene's product activity.
The FKRP knockout cell line enabled the development of a potency assay that is directly linked to the fundamental MoA of the therapy—the restoration of α-DG glycosylation. This functional relevance is a key regulatory requirement, as emphasized by the FDA and EMA [96] [95]. The use of a high-throughput, quantitative method like On-Cell Western provides a platform that is suitable for quality control environments, where robustness, reproducibility, and scalability are essential. This methodology could be adapted for other rAAV products where a key measurable functional output can be defined and linked to the transgene's activity.
The successful validation of a potency assay for AAV2-hRPE65v2 (Luxturna) underscores the importance of a systematic approach to validation, focusing on parameters such as precision, accuracy, specificity, and robustness [97]. As shown in Table 2, setting scientifically justified acceptance criteria for each parameter is critical for regulatory acceptance. Furthermore, the use of a well-characterized reference standard is indispensable for calculating relative potency and ensuring consistency across product batches throughout the product's lifecycle [95].
This case study demonstrates a comprehensive and successful strategy for developing a biologically relevant and robust cell-based potency assay for an rAAV-FKRP gene therapy product. The critical success factor was the strategic generation of a CRISPR-Cas9-mediated FKRP knockout muscle cell line, which provided a clean, disease-relevant biological system for quantifying the product's intended biological effect—the restoration of α-DG glycosylation.
The associated On-Cell Western protocol delivers a quantitative, high-throughput, and mechanism-based method for determining the relative potency of rAAV-FKRP batches. This approach not only meets regulatory requirements for product release and stability testing but also provides a template that can be adapted and applied to the development of potency assays for other gene therapy products targeting monogenic disorders. The integration of advanced genome editing with functional cell-based assays represents a powerful paradigm for ensuring the quality, consistency, and efficacy of next-generation biological medicines.
Conclusion
The successful generation of CRISPR-Cas9 knockout cell lines has become a cornerstone of modern biomedical research, enabling unprecedented precision in functional genomics, disease modeling, and therapeutic development. As the field advances, the integration of AI for sgRNA design, the development of novel Cas variants with reduced off-target effects, and optimized delivery methods are set to further enhance efficiency and reproducibility. These advancements are directly translating into more reliable drug discovery pipelines and robust, clinically relevant potency assays for next-generation gene therapies. The future will see an increased focus on editing complex primary and iPSC-derived cells, paving the way for highly personalized medicine and a deeper understanding of human disease pathophysiology.
References
- 1. CRISPR/Cas9 technology in tumor research and drug ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1552741/full]
- 2. CRISPR/Cas9 therapeutics: progress and prospects [https://www.nature.com/articles/s41392-023-01309-7]
- 3. Cornerstones of CRISPR-Cas in drug development and therapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC5459481/]
- 4. CRISPR-Cas9 and Its Bioinformatics Tools: A Systematic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12109910/]
- 5. AI & CRISPR 2025: How AI Is Revolutionizing Genome Editing [https://aichronicle.co/how-artificial-intelligence-is-revolutionizing-precision-genome-editing-in-2025/]
- 6. Recent applications, future perspectives, and limitations of ... [https://www.sciencedirect.com/science/article/pii/S216225312500188X]
- 7. Protocol for Generation of Single-Gene Knockout in Hard- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12245624/]
- 8. Step-by-Step Protocol for Generating CRISPR-Mediated ... [https://www.protocols.io/view/step-by-step-protocol-for-generating-crispr-mediat-dhg433yw.html]
- 9. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 10. Recent applications, future perspectives, and limitations of ... [https://pubmed.ncbi.nlm.nih.gov/40777740/]
- 11. CRISPR for Disease Modeling and Target Discovery [https://www.discoveryontarget.com/17/crispr-target-discovery]
- 12. Optimization of gene knockout approaches and sgRNA ... [https://www.nature.com/articles/s41598-025-24184-4]
- 13. Protocol for Generation of Single-Gene Knockout in Hard- ... [https://pubmed.ncbi.nlm.nih.gov/40655415/]
- 14. Generating and validating CRISPR-Cas9 knock-out cell lines [https://www.abcam.com/en-us/stories/articles/crispr-cas9-knockout-cell-lines]
- 15. Advances in CRISPR Disease Modeling: Cells, Organoids, ... [https://www.synthego.com/blog/crispr-disease-modeling-trends/]
- 16. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 17. CRISPR Therapeutics Highlights Strategic Priorities and ... [https://ir.crisprtx.com/news-releases/news-release-details/crispr-therapeutics-highlights-strategic-priorities-and]
- 18. Knockout Cell Lines Market Outlook 2025-2032 [https://www.intelmarketresearch.com/diagnostic-and-biotech/6016/knockout-cell-lines-2025-2032-128]
- 19. In-Depth Analysis of Gene Knockout Cell Line Construction ... [https://www.linkedin.com/pulse/forecasting-future-in-depth-analysis-gene-knockout-cell-line-bulee]
- 20. Gene Editing Market Size to Surpass USD 42.13 Billion by [https://www.globenewswire.com/news-release/2025/11/04/3179998/0/en/Gene-Editing-Market-Size-to-Surpass-USD-42-13-Billion-by-2034.html]
- 21. Gene Knockout Cell Line Construction Service Market ... [https://www.towardshealthcare.com/insights/gene-knockout-cell-line-construction-service-market-sizing]
- 22. A benchmark comparison of CRISPRn guide-RNA design ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11386-3]
- 23. Top 15 CRISPR Companies in the US that are making ... [https://www.scispot.com/blog/top-15-crispr-companies-in-the-us]
- 24. Precision gene editing: The power of CRISPR-Cas in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590234/]
- 25. Expanding the genetic editing tool kit: ZFNs, TALENs, and ... [https://www.jci.org/articles/view/72992]
- 26. CRISPR-Cas9 vs TALEN vs ZFN: Precision Gene Editing ... [https://synapse.patsnap.com/article/crispr-cas9-vs-talen-vs-zfn-precision-gene-editing-compared]
- 27. Comparing Gene Editing Platforms: CRISPR vs. Traditional ... [https://blog.crownbio.com/comparing-gene-editing-platforms-crispr-vs-traditional-methods]
- 28. CRISPR-Cas9, TALENs and ZFNs - the battle in gene editing [https://www.ptglab.com/news/blog/crispr-cas9-talens-and-zfns-the-battle-in-gene-editing/?srsltid=AfmBOoqJkkzRrIcHUljUVt1hyJgATcoCgBUCtC2DWo5o0V_q8x6-9Ikq]
- 29. TALEN-Interceded Genome Editing in Plants [https://www.mdpi.com/2223-7747/14/19/3024]
- 30. Pros and cons of ZNFs, TALENs, and CRISPR/Cas [https://www.jax.org/news-and-insights/jax-blog/2014/march/pros-and-cons-of-znfs-talens-and-crispr-cas]
- 31. Recent advances in therapeutic gene-editing technologies [https://www.sciencedirect.com/science/article/pii/S152500162500200X]
- 32. Optimizing sgRNA to Improve CRISPR/Cas9 Knockout Efficiency [https://pmc.ncbi.nlm.nih.gov/articles/PMC8640246/]
- 33. A Guide to Efficient CRISPR gRNA Design: Principles and ... [https://www.genscript.com/a-guide-to-efficient-crispr-grna-design-principles-and-design-tools.html]
- 34. sgRNA Sequence Motifs Blocking Efficient CRISPR/Cas9- ... [https://www.sciencedirect.com/science/article/pii/S2211124719300336]
- 35. Optimizing sgRNA structure to improve CRISPR-Cas9 ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0846-3]
- 36. Revolutionizing CRISPR technology with artificial intelligence [https://www.nature.com/articles/s12276-025-01462-9]
- 37. From Code to Life: The AI‐Driven Revolution in Genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12376515/]
- 38. Artificial Intelligence for CRISPR Guide RNA Design [https://arxiv.org/html/2508.20130v1]
- 39. AI-powered CRISPR could lead to faster gene therapies ... [https://med.stanford.edu/news/all-news/2025/09/ai-crispr-gene-therapy.html]
- 40. CRISPR-GPT — The Future of Bioengineering [https://ersgenomics.com/crispr-gpt-the-future-of-bioengineering/]
- 41. CRISPR Transfection Protocols Guide: How To Select The ... [https://www.synthego.com/guide/how-to-use-crispr/transfection-protocols/]
- 42. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 43. CRISPR/Cas9 Delivery Systems to Enhance Gene Editing ... [https://www.mdpi.com/1422-0067/26/9/4420]
- 44. Optimizing the delivery of CRISPR/Cas9 ... [https://pubmed.ncbi.nlm.nih.gov/40780629/]
- 45. Optimizing the delivery of CRISPR/Cas9 ... [https://www.sciencedirect.com/science/article/abs/pii/S0378111925005049]
- 46. Optimizing CRISPR methodology for precise gene editing ... [https://haematologica.org/article/view/12023]
- 47. Optimization of gene knockout approaches and sgRNA ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12627716/]
- 48. Challenges in lentiviral vector production: retro- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12159608/]
- 49. Generating Single Cell–Derived Knockout Clones in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6741428/]
- 50. Comparing transfection methods for enhanced gene delivery [https://journals.gen.tr/index.php/jsp/article/view/2717]
- 51. Enhancing non-viral gene delivery to human T cells ... [https://www.sciencedirect.com/science/article/pii/S0753332225000149]
- 52. A systematic comparison of in-house prepared transfection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12603952/]
- 53. Protocol for high-efficiency generation of iPSCs stably ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12272859/]
- 54. Characterizing and controlling CRISPR repair outcomes in ... [https://www.nature.com/articles/s41467-025-66058-3]
- 55. Applications of multiplexed CRISPR–Cas for genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322214/]
- 56. Application of a multiplex CRISPR/Cas9 strategy for ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1633104/full]
- 57. Multiplex CRISPR-Cas9 [https://horizondiscovery.com/en/applications/gene-editing/multiplexed-gene-knockout]
- 58. Troubleshooting Low Knockout Efficiency in CRISPR ... [https://www.biosynsis.com/blog/troubleshooting-low-knockout-efficiency-in-crispr-experiments/]
- 59. Limitations of qPCR detection in evaluating CRISPR/Cas9 ... [https://www.genuinbiotech.com/post/limitations-of-qpcr-detection-in-evaluating-crispr-cas9-gene-knockout-efficiency]
- 60. Optimization of the Genome Editing CRISPR-Cas9 ... [https://pubmed.ncbi.nlm.nih.gov/41023259/]
- 61. Optimized protocol to create deletion in adherent cell lines ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8551220/]
- 62. Optimization strategy - Nucleofector ® Technology [https://bioscience.lonza.com/lonza_bs/CH/en/transfection-optimization-strategy-for-the-nucleofector-technology]
- 63. Choosing the Right Transfection Reagent for Optimal ... [https://www.promega.com/resources/pubhub/tpub-205-choosing-the-right-transfection-reagent-for-optimal-efficiency/]
- 64. In Vitro Cell Transfection Reagent Product Selection Guide [https://www.yeasenbio.com/blogs/cell/in-vitro-cell-transfection-reagent-product-selection-guide]
- 65. Rapid Assessment of CRISPR Transfection Efficiency and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8978543/]
- 66. Factors Influencing Transfection Efficiency [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/transfection-basics/factors-influencing-transfection-efficiency.html]
- 67. High-fidelity CRISPR-Cas9 variants with undetectable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4851738/]
- 68. Guide-specific loss of efficiency and off-target reduction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10055116/]
- 69. SpCas9-HF1 enhances accuracy of cell cycle-dependent ... [https://www.sciencedirect.com/science/article/pii/S2162253124000118]
- 70. Optimized CRISPR guide RNA design for two high-fidelity ... [https://www.nature.com/articles/s41467-019-12281-8]
- 71. Off-target effects in CRISPR/Cas9 gene editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10034092/]
- 72. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 73. The hidden risks of CRISPR/Cas: structural variations and ... [https://www.nature.com/articles/s41467-025-62606-z]
- 74. Latest Developed Strategies to Minimize the Off-Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7407193/]
- 75. Targeting DNA repair mechanisms in cancer therapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC12582025/]
- 76. DNA damage repair: historical perspectives, mechanistic ... [https://www.nature.com/articles/s41392-021-00648-7]
- 77. Weaponizing CRISPR/Cas9 for selective elimination of ... [https://www.sciencedirect.com/science/article/pii/S1568786425000369]
- 78. Off-target Effects in CRISPR/Cas9-mediated Genome ... [https://www.sciencedirect.com/science/article/pii/S216225311630049X]
- 79. Systematic Comparison of Computational Tools for Sanger ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10854981/]
- 80. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 81. Next-Generation Sequencers vs. Sanger ... [https://www.labx.com/resources/next-generation-sequencers-vs-sanger-sequencers-key-differences/5597]
- 82. Next-Generation Sequencing (NGS) vs. Sanger ... [https://www.labmanager.com/next-generation-sequencing-ngs-vs-sanger-sequencing-which-dna-sequencing-method-is-right-for-you-33659]
- 83. From Sanger to Oxford Nanopore MinION Technology [https://pmc.ncbi.nlm.nih.gov/articles/PMC12153771/]
- 84. Antibody validation for Western blot: By the user, for the user [https://pmc.ncbi.nlm.nih.gov/articles/PMC6983856/]
- 85. How and why you should validate your antibodies [https://azurebiosystems.com/blog/quantitative-western-blotting-how-and-why-you-should-validate-your-antibodies/]
- 86. A systematic approach to quantitative Western blot analysis [https://www.sciencedirect.com/science/article/pii/S000326971930750X]
- 87. Validation of RNAi Knockdown Using Multiple Reaction ... [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/cell-culture-and-cell-culture-analysis/transfection-and-gene-editing/validation-rnai-knockdown-using-multiple-reaction-monitoring?srsltid=AfmBOoq9ahV5Q_2lvD_8UqRa7xyggRC8vs3hg1l6KDm8l8plG_Y-gS6W]
- 88. How to Validate a CRISPR Knockout [https://biognosys.com/how-to-validate-crispr-knockout/]
- 89. Functional phenotyping of genomic variants using joint ... [https://www.nature.com/articles/s41592-025-02805-0]
- 90. Functional phenotyping of genomic variants using joint ... [https://pubmed.ncbi.nlm.nih.gov/40890552/]
- 91. The CellPhe toolkit for cell phenotyping using time-lapse ... [https://www.nature.com/articles/s41467-023-37447-3]
- 92. Perturbomics: CRISPR–Cas screening-based functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322284/]
- 93. Bioinformatics approaches to analyzing CRISPR screen data [https://pmc.ncbi.nlm.nih.gov/articles/PMC10019185/]
- 94. Recent advances in CRISPR-based functional genomics ... [https://www.nature.com/articles/s12276-024-01212-3]
- 95. Building a Robust Biological Assay for Potency Measurement [https://www.bioprocessintl.com/product-characterization/building-a-robust-biological-assay-for-potency-measurement]
- 96. CRISPR-Cas9 KO Cell Line Generation and Development ... [https://www.mdpi.com/2073-4409/12/20/2444]
- 97. Development and Validation of Cell-Based Potency Assays ... [https://www.marinbio.com/development-and-validation-of-cell-based-potency-assays-for-aav-therapies-insights-from-a-commercial-aav-vector-study/]
- 98. Streamlined Generation of CRISPR/Cas9-Mediated Single- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11091971/]
- 99. An efficient and precise method for generating knockout ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7708952/]