CRISPR-Cas9 Double-Strand Break Formation: Molecular Mechanisms, Applications, and Clinical Translation
This article provides a comprehensive analysis of the CRISPR-Cas9 mechanism for double-strand break (DSB) formation, tailored for researchers, scientists, and drug development professionals.
CRISPR-Cas9 Double-Strand Break Formation: Molecular Mechanisms, Applications, and Clinical Translation
Abstract
This article provides a comprehensive analysis of the CRISPR-Cas9 mechanism for double-strand break (DSB) formation, tailored for researchers, scientists, and drug development professionals. It explores the fundamental structural biology of the Cas9-guide RNA complex and its programmable DNA targeting, detailing the precise molecular events from PAM recognition through R-loop formation to dual-nuclease cleavage. The content covers advanced therapeutic applications across genetic disorders, cancer, and infectious diseases, including recently approved therapies and ongoing clinical trials. It addresses critical challenges including off-target effects, delivery limitations, and repair pathway control, while comparing CRISPR-Cas9 with alternative gene-editing platforms. The synthesis provides a roadmap for optimizing precision editing tools and translating mechanistic insights into clinical breakthroughs.
The Molecular Architecture of CRISPR-Cas9: Deconstructing the DNA Targeting Machinery
Cas9 Protein Architecture and Catalytic Mechanism
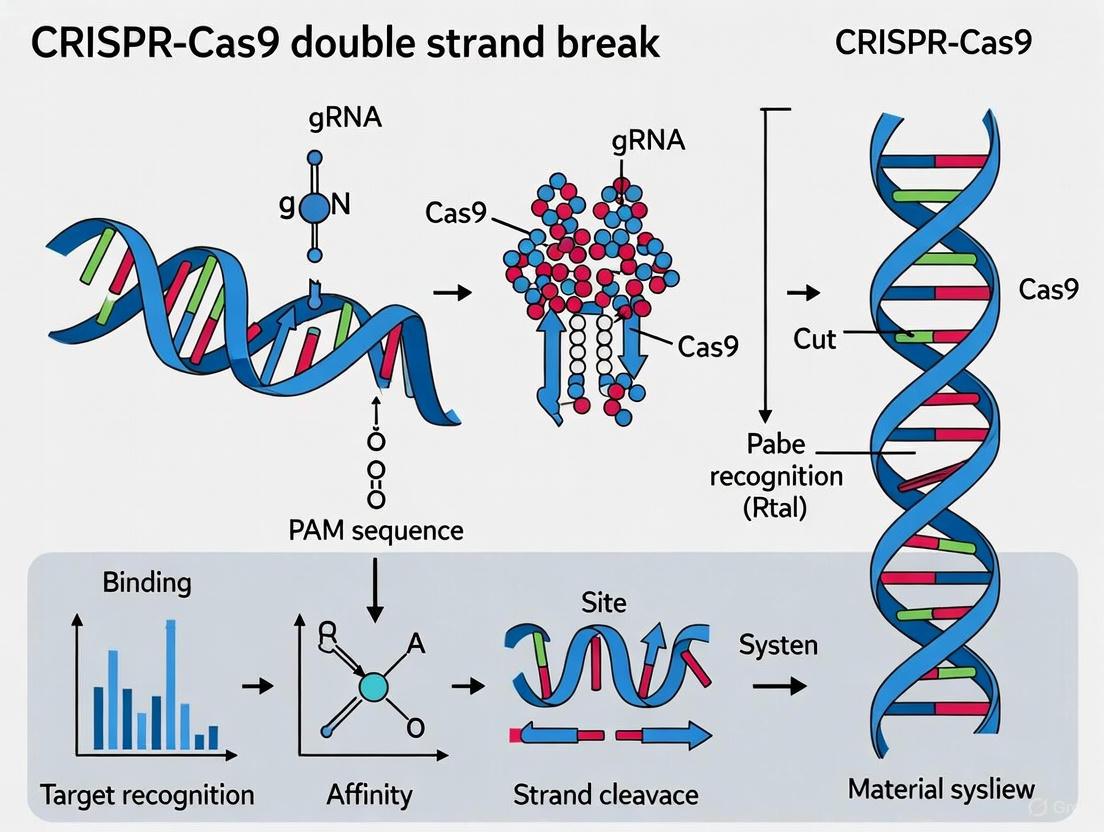
The CRISPR-associated protein 9 (Cas9) is an RNA-guided DNA endonuclease that serves as the central effector molecule in type II CRISPR-Cas systems. Its structure and activation mechanism provide the foundation for its genome-editing capabilities [1] [2].
Structural Organization and Domains
Cas9 exhibits a bilobed architecture composed of two primary lobes: the nuclease (NUC) lobe and the recognition (REC) lobe, connected by two linking segments [1]. The protein encompasses several critical domains and regions:
- RuvC Domain: Located in the NUC lobe, this domain cleaves the non-target DNA strand (the strand not complementary to the guide RNA). It forms a structural core consisting of a six-stranded β sheet surrounded by four α helices [1] [2].
- HNH Domain: This domain is responsible for cleaving the target DNA strand (the strand complementary to the guide RNA). In the apo-Cas9 structure (without bound nucleic acids), the HNH active site is often poorly ordered, suggesting conformational flexibility that becomes ordered upon DNA binding [1] [2].
- PAM-Interacting (PI) Domain: Situated within the NUC lobe, this domain recognizes the protospacer adjacent motif (PAM), a short DNA sequence adjacent to the target site that is essential for self versus non-self discrimination in bacterial immunity [2].
- REC Lobe (REC1, REC2, and REC3 domains): This lobe is primarily responsible for binding the guide RNA and facilitating the recognition of target DNA [2].
Table 1: Core Functional Domains of Streptococcus pyogenes Cas9 (SpCas9)
| Domain/Region | Primary Function | Structural Features |
|---|---|---|
| RuvC Domain | Cleaves the non-complementary DNA strand | Six-stranded β sheet core surrounded by α helices |
| HNH Domain | Cleaves the complementary DNA strand | β-β-α fold, undergoes conformational activation |
| REC Lobe | Facilitates guide RNA binding and target recognition | Primarily α-helical, interacts with guide RNA |
| PAM-Interacting Domain | Recognizes the NGG protospacer adjacent motif | Mediates initial DNA binding and unwinding |
| Arg-rich Region | Likely mediates nucleic acid binding | Connects the two structural lobes (residues 59-76) |
Conformational Activation and DNA Cleavage Mechanism
Cas9 undergoes significant structural rearrangements to transition from an inactive to a DNA-cleaving enzyme. In its apo state, Cas9 exists in a conformation that is incapable of DNA cleavage. The binding of the guide RNA induces a major reorientation of the structural lobes, forming a central channel where DNA substrates are bound. This RNA-induced activation is a critical step, "implicating guide RNA loading as a key step in Cas9 activation" [1].
The process of target DNA recognition and cleavage follows a defined sequence [2]:
- PAM Recognition: The Cas9-gRNA complex searches the DNA through 3D and 1D diffusion. The PAM-interacting domain first identifies a correct PAM sequence (e.g., 5'-NGG-3' for SpCas9).
- DNA Unwinding: PAM binding triggers the unwinding of the adjacent double-stranded DNA, exposing the seed region near the PAM.
- R-loop Formation: The guide RNA undergoes strand invasion, testing for complementarity with the target DNA strand. If fully complementary, a stable R-loop structure forms, displacing the non-target DNA strand.
- Conformational Activation and Cleavage: Successful R-loop formation triggers a final conformational change in Cas9, positioning the HNH domain to cleave the target strand and the RuvC domain to cleave the non-target strand. The HNH domain cuts the target strand 3 base pairs upstream of the PAM, while the RuvC domain cuts the non-target strand 3-5 base pairs away, typically resulting in a blunt-ended double-strand break [2].
Guide RNA Design Principles
The guide RNA (gRNA) is the targeting component that dictates the specificity of the CRISPR-Cas9 system. Its design is paramount to the success and accuracy of any genome-editing experiment [3] [4].
gRNA Components and Function
The gRNA is a chimeric molecule that combines two natural RNA elements [2] [5]:
- crRNA (CRISPR RNA): This component contains the ~20 nucleotide "spacer" sequence that defines the genomic target through Watson-Crick base pairing.
- tracrRNA (trans-activating crRNA): This structural element binds to the Cas9 protein and is essential for its activation.
In most experimental applications, these two elements are fused into a single-guide RNA (sgRNA) of approximately 100 nucleotides, which retains full functionality [2].
Key Parameters for Efficient gRNA Design
On-Target Efficiency
On-target efficiency predicts how effectively a gRNA will mediate editing at the intended target site. Several algorithm-based scoring methods have been developed from large-scale experimental datasets [3]:
- Rule Set 2 & 3: Developed by Doench et al., these models use machine learning (gradient-boosted regression trees) on data from thousands of gRNAs to predict efficiency. Rule Set 3 is the most current and considers the tracrRNA sequence for improved accuracy [3] [4].
- CRISPRscan: This algorithm is a predictive model based on the activity data of 1,280 gRNAs validated in vivo in zebrafish [3].
- Lindel: A logistic regression model that predicts the likelihood and spectrum of insertions and deletions (indels) resulting from Cas9-mediated cleavage, providing a frameshift ratio prediction [3].
Off-Target Minimization
Minimizing off-target effects is crucial for experimental specificity and therapeutic safety. Key assessment methods include [3]:
- Cutting Frequency Determination (CFD) Score: This scoring matrix, referenced in Doench's 2016 work, is based on the activity of 28,000 gRNAs with single variations. A score below 0.05 (or 0.023 in some applications) is generally considered low risk [3].
- MIT Specificity Score (Hsu-Zhang Score): Developed from data on over 700 gRNA variants with 1-3 mismatches, this score helps identify potential off-target sites across the genome [3].
- Homology Analysis: A genome-wide search for sequences similar to the designed gRNA that also contain a valid PAM. Fewer than three nucleotide mismatches, particularly those far from the PAM, are of concern [3].
Table 2: gRNA Design Considerations for Different Experimental Goals
| Experimental Goal | Primary Design Consideration | Optimal gRNA Location | Key Constraints |
|---|---|---|---|
| Gene Knockout (NHEJ) | gRNA Sequence Efficiency | 5' - 65% of protein-coding region | Avoid very N- or C-terminal to prevent functional truncated proteins [4]. |
| Precise Editing (HDR) | Proximity to Edit | Within ~30 nt of the desired edit [4] | Few gRNA choices; may need alternative Cas enzymes with different PAMs [4]. |
| CRISPRa (Activation) | Location relative to TSS | ~100 nt window upstream of TSS [4] | Accurate TSS annotation (e.g., FANTOM database) is critical [4]. |
| CRISPRi (Inhibition) | Location relative to TSS | ~100 nt window downstream of TSS [4] | Accurate TSS annotation is critical [4]. |
Workflow for gRNA Design and Selection
A robust gRNA selection process involves multiple steps to balance efficiency and specificity [3] [5]:
- Identify PAM Sites: Locate all NGG (for SpCas9) sequences in the target genomic region.
- Define Candidate gRNAs: For each PAM, identify the 20 nucleotides immediately 5' to it as the potential gRNA spacer sequence.
- Score for Efficiency: Use algorithms like Rule Set 3 to rank gRNAs by predicted on-target activity.
- Analyze for Specificity: Perform a genome-wide off-target analysis using CFD or MIT scoring to shortlist gRNAs with minimal off-target risks.
- Final Selection: For gene knockout, select 2-3 high-ranking gRNAs targeting different regions of the gene to control for target accessibility and confirm phenotype consistency [4].
Experimental Protocols for Analyzing Cas9 Function and DSB Repair
Understanding the outcomes of Cas9-induced double-strand breaks requires precise methodologies to quantify editing efficiency and repair dynamics.
Quantifying DSB Dynamics with UMI-DSBseq
A advanced method for characterizing DSB induction and repair is UMI-DSBseq, a molecular and computational toolkit that enables multiplexed quantification of DSB intermediates and repair products by single-molecule sequencing [6].
Key Protocol Steps [6]:
- Delivery: Preassembled Cas9 ribonucleoproteins (RNPs) are delivered directly into cells (e.g., tomato protoplasts) via PEG-mediated transformation to ensure synchronized DSB induction.
- Time-Course Sampling: Cells are harvested at multiple time points (e.g., over 72 hours) post-transformation.
- Library Preparation: Genomic DNA is extracted and subjected to the UMI-DSBseq protocol:
- End Repair: DNA ends are repaired by fill-in of 3' overhangs.
- Adaptor Ligation: Adapters containing Unique Molecular Identifiers (UMIs) are ligated directly to both unrepaired DSBs and to intact molecules (at a flanking restriction enzyme site cleaved in vitro).
- Sequencing: Illumina sequencing-ready libraries are prepared and sequenced.
- Data Analysis: Sequencing reads are categorized into:
- Unrepaired DSBs
- Wild-type intact molecules (precisely repaired or uncut)
- Indel-containing products of error-prone repair
This approach allows researchers to directly measure the rates of Cas9 cutting, precise repair, and error-prone repair, revealing that precise repair can account for up to 70% of all repair events in plant protoplasts [6].
Standard Workflow for CRISPR Gene Knockout
A common CRISPR-Cas9 experiment for gene disruption follows this protocol [5]:
- Design gRNA: Target an early exon of the gene of interest using the principles in Section 2.
- Obtain gRNA:
- Option A (Synthetic): Order a chemically synthesized sgRNA.
- Option B (Plasmid-based): Clone the gRNA sequence into an expression plasmid (e.g., via Gibson Assembly) for delivery.
- Deliver CRISPR Components: Co-deliver Cas9 and gRNA expression constructs or preassembled RNPs into your target cells using system-appropriate methods (e.g., lipofection, electroporation, microinjection).
- Screen and Validate:
- Enrich edited cells via selection (if using a selectable marker).
- Screen candidate clones by PCR amplification of the target region.
- Sequence the PCR products to determine the exact indel sequences and identify homozygous edits.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for CRISPR-Cas9 Genome Editing Experiments
| Reagent / Tool | Function / Description | Example Sources / Notes |
|---|---|---|
| SpCas9 Nuclease | The effector protein that creates DSBs; can be delivered as protein, mRNA, or encoded in a plasmid. | Widely available from commercial suppliers (e.g., IDT, Thermo Fisher). |
| Synthetic sgRNA | Chemically synthesized single-guide RNA; offers immediate activity and avoids cloning steps. | Companies like GenScript, IDT, and Synthego provide synthesis services. |
| CRISPR Plasmids | DNA vectors for in-cell expression of Cas9 and gRNA; suitable for viral packaging (lentivirus, AAV). | Non-profit repositories (e.g., Addgene) are common sources. |
| HDR Donor Templates | Single-stranded oligodeoxynucleotides (ssODNs) or double-stranded DNA templates for precise editing. | For edits <200 nt, use ssODNs; for larger edits, use dsDNA fragments or plasmid donors. |
| gRNA Design Tools | Web-based platforms for designing and scoring gRNAs for on-target efficiency and off-target effects. | CRISPick (Broad Institute), CHOPCHOP, CRISPOR, GenScript Design Tool [3]. |
| Validation Primers | Oligonucleotides for PCR amplification and sequencing of the target locus to confirm edits. | Standard custom oligo synthesis. |
| AI Design Assistants | AI tools that leverage published data to automate experimental design and predict off-targets. | CRISPR-GPT (Stanford) acts as a gene-editing "copilot" for researchers [7]. |
Advanced Applications and Current Research Frontiers
The understanding of Cas9 structure and refinement of gRNA design principles has directly enabled the translation of CRISPR technology into therapeutic applications and advanced research tools.
Clinical Applications and Therapeutic Genome Editing
CRISPR-based therapies have progressed rapidly into clinical trials, with the first medicines receiving approval [8]:
- Casgevy (exagamglogene autotemcel): This therapy, approved for sickle cell disease and transfusion-dependent beta thalassemia, uses ex vivo CRISPR-Cas9 to edit hematopoietic stem cells to reactivate fetal hemoglobin [8].
- In Vivo CRISPR Therapies: Intellia Therapeutics' phase I trial for hereditary transthyretin amyloidosis (hATTR) demonstrated the feasibility of systemic, in vivo CRISPR-Cas9 therapy. The treatment, delivered via lipid nanoparticles (LNPs) that accumulate in the liver, achieved ~90% reduction in disease-related protein levels that was sustained over two years [8].
- Personalized Therapies: A landmark case in 2025 reported the first personalized, on-demand in vivo CRISPR treatment for an infant with CPS1 deficiency. The therapy was developed, approved, and delivered in just six months, establishing a regulatory precedent for bespoke gene therapies [8].
AI-Powered Experimental Design
The complexity of CRISPR experimental design has led to the development of AI tools to assist researchers. CRISPR-GPT, a large language model developed at Stanford Medicine, was trained on 11 years of expert discussions and published scientific papers [7]. It functions as a gene-editing "copilot" that can [7]:
- Generate experimental plans for specific goals (e.g., CRISPR activation).
- Predict potential off-target edits and their likely impact.
- Explain the rationale behind each design step, flattening the learning curve for novice users.
- Incorporate safeguards to prevent the design of unethical experiments (e.g., editing human embryos).
The revolutionary power of CRISPR-Cas9 genome editing is built upon the foundational biology of the Cas9 protein's structure and its guided interaction with target DNA. The bilobed architecture, encompassing nuclease and recognition lobes, undergoes precise conformational changes that are activated by guide RNA binding and culminate in site-specific DNA cleavage. Harnessing this mechanism requires meticulous gRNA design informed by sophisticated algorithms that predict on-target efficiency and minimize off-target effects, with design strategies tailored to specific experimental goals from gene knockout to precise editing. As the field advances, these core principles of protein engineering and guide design are being augmented by AI-driven tools and sophisticated delivery systems, paving the way for an expanding frontier of therapeutic applications and fundamental research discoveries.
Protospacer Adjacent Motif (PAM) recognition serves as the fundamental gateway that enables CRISPR-Cas systems to distinguish between self and non-self DNA, initiating a cascade of events culminating in targeted double-strand break (DSB) formation. This sequence-specific recognition mechanism, while constraining the targetable genomic space, provides the critical first step in DNA interrogation by CRISPR-associated nucleases. Recent structural and biochemical studies have elucidated the sophisticated molecular machinery underlying PAM binding, revealing how Cas nucleases achieve remarkable specificity while navigating the challenges of off-target effects. This technical review examines PAM recognition within the broader context of CRISPR-Cas9 mechanisms for DSB formation research, providing researchers with current methodologies, structural insights, and clinical implications of this pivotal process.
The Protospacer Adjacent Motif (PAM) is a short, specific DNA sequence (typically 2-6 base pairs) adjacent to the target DNA region cleaved by the CRISPR system. This motif represents an essential recognition element that must be identified by the Cas nuclease before it can unwind the DNA and verify complementarity with the guide RNA [9]. In the context of bacterial adaptive immunity, the PAM serves a vital function in self versus non-self discrimination—while foreign viral DNA contains PAM sequences, the bacterial genome itself lacks these motifs adjacent to stored viral sequences in the CRISPR array, thus preventing autoimmunity [9] [10].
From a mechanistic perspective, the PAM is required for Cas nuclease activation and subsequent DSB formation. The most extensively characterized Cas9 from Streptococcus pyogenes (SpCas9) recognizes a 5'-NGG-3' PAM sequence located directly downstream of the target sequence in the genomic DNA [11]. The PAM is not part of the guide RNA sequence but must be present in the target DNA, generally found 3-4 nucleotides downstream from the Cas9 cut site [9]. This strategic positioning enables the PAM to serve as an initial binding site that triggers local DNA melting, allowing the guide RNA to interrogate the adjacent sequence through RNA-DNA pairing [2].
Molecular Mechanisms of PAM Recognition
Structural Basis of PAM Interaction
PAM recognition occurs primarily through specific domains within the Cas nuclease that interact directly with the DNA backbone and nucleobases. Structural studies have revealed that SpCas9 employs a PAM-interacting (PI) domain containing positively charged residues that form specific hydrogen bonds with the nucleobases of the PAM sequence [2] [12]. Specifically, residues R1333 and R1335 in SpCas9 confer specificity for the two guanines in the NGG PAM by forming four hydrogen bonds with their Hoogsteen faces [12].
When the Cas9-gRNA complex searches for target sites, it first binds to PAM sequences through a combination of 3D and 1D diffusion [2]. PAM recognition triggers conformational changes that destabilize the adjacent DNA duplex, facilitating initial unwinding of the seed region (the PAM-proximal portion of the target sequence) and enabling the guide RNA to initiate strand invasion [2]. This process leads to the formation of an R-loop structure—a triple-stranded intermediate comprising the RNA-DNA hybrid and displaced non-target DNA strand [2].
PAM-Dependent Activation Cascade
The molecular events following PAM recognition occur in a defined sequence:
- Initial PAM Binding: The Cas nuclease scans DNA for compatible PAM sequences through superficial contacts with the DNA backbone [10] [12].
- DNA Destabilization: PAM binding induces conformational changes in the Cas protein that destabilize the adjacent DNA duplex, typically melting 4-5 base pairs upstream of the PAM [2].
- Seed Region Interrogation: The guide RNA tests complementarity with the exposed single-stranded DNA in the seed region [2].
- R-loop Propagation: If seed pairing is successful, the R-loop extends through the entire target region, with full complementarity triggering nuclease activation [2] [12].
- DSB Formation: The HNH nuclease domain cleaves the target strand 3 base pairs upstream of the PAM, while the RuvC domain cleaves the non-target strand 3-5 base pairs away [2].
Table 1: Key Domains Involved in PAM Recognition and DNA Cleavage
| Domain | Function | Specific Role in PAM Recognition |
|---|---|---|
| PAM-Interacting (PI) Domain | PAM recognition and binding | Contains residues that directly contact PAM nucleobases; determines PAM specificity |
| REC Lobe | Guide RNA handling and DNA hybridization | Facilitates DNA melting after PAM recognition; stabilizes R-loop formation |
| HNH Domain | Target strand cleavage | Activated upon complete R-loop formation; cleaves 3 bp upstream of PAM |
| RuvC Domain | Non-target strand cleavage | Cleaves non-target strand after conformational changes triggered by PAM binding |
PAM Diversity Across CRISPR Systems
The requirement for a specific PAM sequence varies considerably among different Cas nucleases, presenting both constraints and opportunities for genome engineering applications. While SpCas9 recognizes a 5'-NGG-3' PAM, other Cas nucleases exhibit distinct PAM preferences, enabling targeting of different genomic regions [9].
Table 2: PAM Sequences for Various Cas Nucleases Used in CRISPR Experiments
| CRISPR Nuclease | Organism Isolated From | PAM Sequence (5' to 3') |
|---|---|---|
| SpCas9 | Streptococcus pyogenes | NGG |
| hfCas12Max | Engineered from Cas12i | TN and/or TNN |
| SaCas9 | Staphylococcus aureus | NNGRR(N) |
| NmeCas9 | Neisseria meningitidis | NNNNGATT |
| CjCas9 | Campylobacter jejuni | NNNNRYAC |
| LbCas12a (Cpf1) | Lachnospiraceae bacterium | TTTV |
| AacCas12b | Alicyclobacillus acidiphilus | TTN |
| Cas3 | in silico analysis of various prokaryotic genomes | No PAM requirement |
The diversity of PAM requirements has significant implications for experimental design. When a target genomic locus lacks the preferred PAM for a given nuclease, researchers can select an alternative Cas protein with a compatible PAM [9]. Furthermore, protein engineering approaches have generated Cas variants with altered PAM specificities, including near-PAMless versions such as SpRY-Cas9, which dramatically expand the targetable genome [12].
Experimental Approaches for PAM Identification
Several methodological approaches have been developed to identify and characterize PAM sequences for both natural and engineered Cas nucleases. These techniques range from computational predictions to high-throughput experimental screens.
In Silico Prediction Methods
Initial PAM identification often relies on bioinformatic analysis of protospacer sequences adjacent to CRISPR spacers. Tools such as CRISPRTarget and CRISPRFinder can extract spacer sequences and identify conserved PAM motifs through alignment-based approaches [10]. While computationally efficient, these methods depend on the availability of sequenced phage genomes and cannot distinguish between spacer acquisition motifs (SAMs) and target interference motifs (TIMs) [10].
High-Throughput Experimental Screens
Several experimental approaches have been developed for comprehensive PAM characterization:
Plasmid Depletion Assays: These assays introduce randomized DNA stretches adjacent to target sequences within plasmids transformed into hosts with active CRISPR-Cas systems. Plasmids with "inactive" PAMs are retained and identified via next-generation sequencing, revealing functional PAM elements through depletion patterns [10].
PAM-SCANR (PAM Screen Achieved by NOT-gate Repression): This high-throughput in vivo method utilizes catalytically dead Cas9 (dCas9) fused to a repressor domain. When dCas9 binds to a functional PAM, it represses GFP expression. Fluorescence-activated cell sorting (FACS), plasmid purification, and sequencing then identify functional PAM motifs [10].
In Vitro Cleavage Assays: These approaches use purified Cas effector complexes to cleave DNA libraries containing randomized PAM sequences. Positive screening sequences enriched cleavage products, while negative screening sequences all remaining uncleaved targets [10]. These methods allow for larger library coverage and better control over reaction conditions but require purified, stable effector complexes [10].
Diagram 1: Experimental Workflow for PAM Identification. This flowchart illustrates the major approaches for determining PAM specificity, combining computational and experimental methods.
PAM Engineering and PAMless Variants
Recent protein engineering efforts have focused on reducing PAM restrictions to expand the targetable genome. Directed evolution and structure-based engineering have produced Cas9 variants with altered PAM specificities, with SpRY-Cas9 representing the most promiscuous variant [12].
SpRY-Cas9 contains multiple mutations (R1333P, R1335Q, A61R, L1111R, D1135L, S1136W, G1218K, E1219Q, N1317R, A1322R, and T1337R) that collectively enable recognition of virtually any PAM sequence [12]. Structural analyses reveal that SpRY achieves PAM flexibility through conformational adaptability within its PAM-interacting region, forming non-specific electrostatic interactions with the DNA backbone rather than specific base contacts [12].
However, this PAM flexibility comes with functional trade-offs. Single-molecule studies demonstrate that while SpRY binds target sequences with similar affinity to wild-type Cas9, it exhibits prolonged binding to off-target sites, resulting in slower target identification and increased potential for off-target effects [12]. The mechanism of PAMless recognition involves:
- Backbone interactions: SpRY forms extensive non-specific contacts with the phosphodiester backbone of target DNA
- Conformational flexibility: Solvent-exposed residues adopt different rotamers to accommodate diverse PAM sequences
- Reduced interrogation speed: SpRY cleaves target DNA approximately 1000-fold slower than wild-type Cas9
Research Reagent Solutions for PAM Studies
Table 3: Essential Research Tools for Investigating PAM Recognition
| Reagent/Tool | Function | Application Notes |
|---|---|---|
| SpCas9 Nuclease | Gold standard for PAM recognition studies | Requires 5'-NGG-3' PAM; widely characterized |
| SpRY-Cas9 | Near-PAMless variant for expanded targeting | Useful when target lacks canonical PAM; higher off-target potential |
| PAM-SCANR System | High-throughput PAM identification | Enables comprehensive PAM profiling in vivo |
| dCas9 Variants | Catalytically inactive Cas9 for binding studies | Useful for visualizing target search without cleavage |
| Structural Biology Tools | Cryo-EM, X-ray crystallography | Elucidate molecular mechanisms of PAM recognition |
| Single-Molecule Imaging | DNA curtains, fluorescence microscopy | Visualize real-time target search and binding dynamics |
Clinical Implications and Safety Considerations
PAM recognition has direct implications for therapeutic genome editing applications. While relaxing PAM requirements expands potential therapeutic targets, it also introduces safety considerations. Off-target effects remain a primary concern for clinical translation, as Cas9 can tolerate mismatches between the guide RNA and DNA target, particularly in PAM-distal regions [13].
Recent studies have revealed that CRISPR editing can induce large structural variations (SVs), including chromosomal translocations and megabase-scale deletions, beyond the well-characterized small indels [14]. These SVs raise substantial safety concerns for clinical applications and highlight the importance of comprehensive off-target assessment [14].
Strategies to mitigate off-target effects while maintaining efficient on-target editing include:
- High-fidelity Cas variants: Engineered Cas9 variants with enhanced specificity, such as HiFi Cas9 [13]
- Paired nickase systems: Using two Cas9 nickases to create adjacent single-strand breaks instead of a DSB [13]
- Computational guide design: Careful selection of guide sequences with minimal off-target potential using tools like Cas-OFFinder [13]
- Delivery optimization: Controlling Cas9 expression levels and duration to limit off-target exposure [8]
The ongoing clinical development of CRISPR-based therapies, including the recently approved Casgevy for sickle cell disease and beta thalassemia, underscores the critical importance of understanding PAM recognition and its relationship to editing specificity [8] [15]. As of February 2025, over 150 active clinical trials are investigating CRISPR-based therapies across numerous disease areas, making the optimization of PAM recognition and target specificity more relevant than ever [15].
PAM recognition represents the critical initial step in CRISPR-mediated genome editing, serving as the gateway to target site specificity. Understanding the molecular mechanisms underlying PAM binding, the diversity of PAM requirements across Cas nucleases, and the experimental approaches for characterizing PAM interactions provides researchers with the foundation necessary for designing precise genome editing experiments. While recent engineering efforts have created Cas variants with relaxed PAM requirements, these advances come with trade-offs in specificity and kinetics that must be carefully considered in both basic research and therapeutic applications. As CRISPR technology continues to evolve, the relationship between PAM recognition and editing outcomes will remain a central consideration for achieving specific and safe genome modification.
DNA interrogation represents the critical initial phase in the CRISPR-Cas mediated genome editing workflow, during which the Cas nuclease identifies and verifies its target DNA sequence prior to cleavage. This process encompasses two fundamental steps: protospacer adjacent motif (PAM) recognition and DNA unwinding to facilitate guide RNA-DNA hybridization, culminating in the formation of a three-stranded structure known as the R-loop [16] [17]. The efficiency and fidelity of this interrogation process directly determine the overall success and specificity of subsequent genome editing outcomes. Within the context of CRISPR-Cas9 mechanism research, understanding DNA interrogation is paramount for elucidating how double-strand breaks are initiated and controlled. This technical guide provides an in-depth examination of the molecular mechanics underlying DNA interrogation, synthesizing recent structural and biophysical findings to present a coherent model of target recognition from initial scanning to stable R-loop formation, with particular emphasis on implications for double-strand break formation research.
Molecular Mechanics of Target Recognition
PAM Recognition and Specificity
The CRISPR-Cas system initiates DNA interrogation through identification of a short protospacer adjacent motif (PAM) flanking the target sequence. This step serves as an essential initial checkpoint that distinguishes self from non-self DNA, thereby preventing autoimmune targeting of the bacterial CRISPR locus [17] [18]. For Streptococcus pyogenes Cas9 (SpyCas9), the most extensively characterized Cas nuclease, the PAM sequence consists of a 5'-NGG-3' motif, where "N" represents any nucleotide [17]. PAM recognition is mediated through specific protein-DNA interactions that trigger conformational changes in the Cas complex, priming it for subsequent DNA unwinding.
Recent research reveals a fundamental trade-off between PAM-binding specificity and genome-editing efficiency. SpyCas9 variants with reduced PAM specificity demonstrate persistent non-selective DNA binding and recurrent failures to engage target sequences through stable guide RNA hybridization, ultimately leading to reduced editing efficiency in cellular environments [16]. This suggests that efficient editing is favored by specific yet weak PAM binding coupled with rapid DNA unwinding, rather than broad PAM recognition capabilities [16].
Table 1: PAM Requirements and Recognition Mechanisms Across CRISPR Systems
| CRISPR System | PAM Sequence | Recognition Mechanism | Specificity Considerations |
|---|---|---|---|
| SpyCas9 | 5'-NGG-3' | Protein-DNA interactions via PI domain | High specificity required for efficient editing |
| Cas12a | 5'-TTTN-3' | Protein-DNA interactions | T-rich PAM enables targeting of distinct genomic regions |
| Type I-E Systems | Promiscuous recognition | Multi-subunit Cascade complex | Broader PAM recognition with reduced specificity |
DNA Unwinding and R-loop Formation Trajectory
Following PAM recognition, Cas nucleases initiate DNA unwinding to permit guide RNA hybridization with the target DNA strand. This process proceeds through a defined sequence of intermediates that have been characterized through sophisticated biophysical techniques. Single-molecule torque spectroscopy studies reveal that Cas12a orthologs engage target DNA through a multi-step pathway marked by distinct REC domain arrangements [19] [20].
The R-loop formation initiates with the generation of a seed bubble at the PAM-proximal region, involving unwinding of approximately 11 base pairs of dsDNA to scout for sequence complementarity [18]. This is followed by directional propagation of the R-loop toward the PAM-distal end, ultimately resulting in a full R-loop structure where approximately 20 base pairs of the guide RNA-DNA heteroduplex are formed [18]. Throughout this process, the non-target DNA strand is displaced, creating the characteristic three-stranded R-loop structure.
High-resolution structural studies of Type I-E Cascade systems reveal that PAM recognition induces severe DNA bending, leading to spontaneous DNA unwinding that nucleates from the seed sequence [18]. Cryo-EM snapshots of Thermobifida fusca Type I-E Cascade captured at different stages of R-loop formation show that initial PAM binding causes DNA bending, which in turn facilitates spontaneous DNA unwinding to form the seed-bubble intermediate [18].
Comparative Analysis of Cas9 and Cas12a Interrogation Mechanisms
Kinetic Intermediates and Conformational Transitions
The DNA interrogation pathways of Cas9 and Cas12a, while sharing fundamental similarities, exhibit distinct kinetic intermediates and conformational transitions. Single-molecule studies utilizing gold rotor bead tracking (AuRBT) have enabled direct observation of these intermediates at base-pair resolution under biologically relevant supercoiling conditions [19].
For Cas9, R-loop formation proceeds through a discrete intermediate corresponding to its approximately 9 bp seed region, with DNA supercoiling strongly modulating activity and specificity by controlling R-loop dynamics [19]. In contrast, Cas12a exhibits a more complex multi-step pathway with distinct intermediates, including a ~5 bp seed intermediate and a ~17 bp intermediate that likely represents a pre-cleavage conformation [19]. These intermediates display ortholog-dependent characteristics, with Acidaminococcus sp. Cas12a (AsCas12a) showing clear dwells in the ~5 bp intermediate during R-loop formation and collapse, a feature not observed in Lachnospiraceae bacterium Cas12a (LbCas12a) under identical conditions [19].
Table 2: Kinetic Intermediates in Cas9 and Cas12a R-loop Formation
| Parameter | Cas9 | Cas12a |
|---|---|---|
| Seed Intermediate Size | ~9 bp | ~5 bp |
| Secondary Intermediate | Not observed | ~17 bp |
| Downstream Unwinding | Limited | Transient "breathing" beyond R-loop |
| Supercoiling Sensitivity | High | Ortholog-dependent |
| Mismatch Tolerance | Increased on underwound DNA | Varies by ortholog |
Structural Determinants of Interrogation Fidelity
The structural features governing DNA interrogation fidelity differ significantly between Cas9 and Cas12a systems. Cas12a possesses dramatic domain flexibility that limits protein-DNA contacts until nearly complete R-loop formation, with distinct REC domain arrangements marking stages of R-loop formation [20]. This flexibility prevents premature nuclease activation, as the non-target strand is only pulled across the RuvC nuclease when domain docking occurs after extensive R-loop formation [20].
For Cas9, the two-step target capture mechanism involves initial PAM binding followed by DNA unwinding, with the balance between these steps crucial for editing efficiency [16]. Reduced PAM specificity creates kinetic traps that slow both target search and unwinding dynamics, ultimately diminishing genome-editing efficiency [16]. This mechanistic understanding has led to engineering strategies aimed at optimizing the two-step process for enhanced editing performance.
Experimental Methodologies for Studying DNA Interrogation
Single-Molecule Biophysical Approaches
Advanced single-molecule techniques have revolutionized our ability to probe DNA interrogation dynamics with unprecedented temporal and spatial resolution. Gold rotor bead tracking (AuRBT), a derivative of magnetic tweezers, enables direct measurement of R-loop formation at base-pair resolution under controlled DNA supercoiling conditions [19]. This methodology involves constraining a DNA tether containing a PAM and target sequence between a cover glass and a paramagnetic bead, while a gold nanoparticle attached to the DNA side is tracked at high speed to measure torque and twist changes associated with R-loop formation [19].
Single-molecule FRET (smFRET) provides complementary insights into conformational dynamics during DNA interrogation, particularly through monitoring distance changes between fluorescently labeled protein and DNA components. When combined with magnetic tweezers, these approaches can simulate cellular environments by applying supercoiling to DNA targets, replicating the topological stress encountered in physiological conditions [19]. The experimental workflow typically involves:
- DNA construct preparation with target sequences and flanking handles for attachment
- Surface immobilization of DNA molecules in flow chambers
- Protein and guide RNA introduction under controlled buffer conditions
- Data acquisition during supercoiling cycles to induce R-loop formation and collapse
- Change-point analysis to identify discrete transitions between interrogation states
Figure 1: Experimental Workflow for Single-Molecule DNA Interrogation Studies
Structural Biology Techniques
High-resolution structural biology methods have provided invaluable insights into the molecular architecture of CRISPR-DNA complexes during interrogation. Cryo-electron microscopy (cryo-EM) has emerged as a particularly powerful approach, enabling visualization of transient intermediates along the R-loop formation pathway [18] [20]. Recent advances in cryo-EM have allowed researchers to capture wild-type Cas12a at various stages of R-loop formation and DNA delivery into the RuvC active site, revealing how domain flexibility guides the interrogation process [20].
The standard protocol for structural studies of DNA interrogation includes:
- Complex reconstitution by incubating Cas protein, guide RNA, and target DNA
- Vitrification through rapid freezing in liquid ethane
- Data collection using high-end cryo-electron microscopes
- Image processing and 3D reconstruction
- Model building and refinement into density maps
These structural approaches have revealed that Cas12a R-loop formation initiates from a 5-bp seed, with distinct REC domain arrangements marking progressive stages of R-loop formation [20]. Similarly, studies of Type I-E Cascade have captured structural snapshots of seed-bubble formation and full R-loop assembly, providing temporal and spatial resolution of key mechanistic steps [18].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for DNA Interrogation Studies
| Reagent / Tool | Function / Application | Technical Considerations |
|---|---|---|
| dCas9/dCas12a (Nuclease-deficient) | Enables R-loop studies without cleavage | Permits long, repeated measurements on single DNA molecules |
| Site-specifically labeled DNA constructs | Single-molecule visualization | Typically includes biotin/ligand handles for surface attachment |
| Gold nanoparticles (for AuRBT) | Torque and twist measurement | ~100nm particles attached to DNA for high-resolution tracking |
| Modified guide RNAs (fluorescently labeled) | FRET-based conformational monitoring | Fluorophore positioning critical for signal optimization |
| Supercoiled DNA substrates | Mimicking cellular DNA topology | Prepared through ligation and enzyme treatment |
| UMI-DSBseq toolkit | Quantifying DSB intermediates and repair products | Enables single-molecule resolution of repair dynamics [6] |
Implications for Double-Strand Break Formation Research
The mechanistic insights gleaned from DNA interrogation studies have profound implications for understanding and controlling double-strand break formation in CRISPR-based applications. Recent research utilizing the UMI-DSBseq toolkit for multiplexed quantification of DSB intermediates and repair products has revealed that 64-88% of target molecules are cleaved across three endogenous loci analyzed in tomato protoplasts, while indels resulting from error-prone repair ranged between 15-41% [6]. This significant discrepancy between cleavage and mutagenic repair rates highlights the substantial role of precise repair in determining final editing outcomes.
Kinetic modeling of DSB induction and repair dynamics suggests that indel accumulation is determined by the combined effect of DSB induction rates, processing of broken ends, and the balance between precise versus error-prone repair [6]. Precise repair accounts for most of the gap between cleavage and error repair, representing up to 70% of all repair events at certain targets [6]. These findings underscore how the fidelity of DNA interrogation and subsequent repair processes collectively shape the efficiency of CRISPR-mediated mutagenesis.
The influence of cellular factors on DSB repair further modulates editing outcomes. PARP1, a key DNA damage response protein, has been identified as a significant regulator of repair pathway choice following CRISPR-Cas9 induced DSBs [21]. PARP1 downregulation increases both NHEJ and MMEJ repair without altering homologous recombination, while PARP1 overexpression reduces NHEJ and HR efficiency [21]. This suggests that targeted modulation of DNA repair factors could provide a strategy for biasing DSB repair outcomes toward more predictable or precise edits.
DNA interrogation represents a sophisticated molecular process that governs the specificity and efficiency of CRISPR-mediated genome editing. The two-step mechanism of PAM recognition followed by directional R-loop formation through discrete intermediates ensures targeted DNA binding, while conformational transitions in Cas nucleases license subsequent nuclease activation. Single-molecule biophysical approaches and high-resolution structural biology have collectively illuminated the dynamic nature of this process, revealing how kinetic intermediates and cellular factors influence editing outcomes. Understanding DNA interrogation mechanics provides a fundamental framework for developing next-generation CRISPR tools with enhanced precision and efficacy, particularly for therapeutic applications requiring predictable double-strand break formation and repair. As research in this field advances, continued refinement of our DNA interrogation models will undoubtedly yield new insights into the complex interplay between target recognition, cleavage activation, and DNA repair in diverse genomic contexts.
The CRISPR-Cas9 system has emerged as a revolutionary tool for genome editing, with its core functionality relying on the formation of precise double-strand breaks (DSBs) in DNA. The Cas9 endonuclease achieves this through the coordinated action of two distinct nuclease domains: the HNH and RuvC-like domains [22]. These domains operate via different mechanistic principles to cleave the two strands of the target DNA. The HNH domain cleaves the DNA strand complementary to the CRISPR RNA (crRNA) guide sequence, while the RuvC-like domain cleaves the non-complementary strand [22] [23]. Understanding the precise molecular mechanisms of these domains is fundamental to advancing CRISPR-based research and therapeutic applications, as it informs the development of more precise editing tools and helps mitigate challenges such as off-target effects.
Molecular Mechanisms of the HNH and RuvC Nuclease Domains
The HNH Nuclease Domain
The HNH domain is characterized by a ββα-metal fold and is responsible for cleaving the crRNA-complementary strand of the target DNA. This domain functions through a fixed-position cleavage mechanism [22]. Structural studies indicate that the HNH domain undergoes a large conformational rearrangement upon target DNA binding, positioning itself to catalyze cleavage at a specific phosphodiester bond located 3 base pairs upstream of the protospacer adjacent motif (PAM) [22]. The catalytic core of the HNH domain typically contains conserved histidine and asparagine residues that coordinate a metal ion (most commonly Mg²⁺) essential for hydrolyzing the DNA backbone [24].
The RuvC-like Nuclease Domain
The RuvC-like domain, which shares structural homology with the RNase H superfamily of nucleases, cleaves the DNA strand non-complementary to the crRNA. In contrast to the HNH domain, it employs a ruler-based cleavage mechanism [22]. This domain measures a fixed distance from the PAM sequence to determine its cleavage site, typically resulting in a cut 3-8 nucleotides upstream of the PAM [22]. The RuvC active site utilizes a DED catalytic motif that coordinates two metal ions to facilitate DNA cleavage, a mechanism conserved across the RNase H superfamily [25] [26].
Table 1: Comparative Analysis of HNH and RuvC Nuclease Domains in Cas9
| Feature | HNH Domain | RuvC-like Domain |
|---|---|---|
| Biological Origin | Widespread nuclease family found in colicins, homing endonucleases, and phage packaging proteins (e.g., HK97 gp74) [24] | Bacterial resolvase involved in Holliday junction resolution during DNA repair [25] |
| Primary Function in Cas9 | Cleaves the target DNA strand complementary to the crRNA guide sequence [22] | Cleaves the target DNA strand non-complementary to the crRNA guide sequence [22] |
| Cleavage Mechanism | Fixed position relative to the PAM sequence [22] | Ruler mechanism measuring distance from the PAM [22] |
| Catalytic Motif/Residues | Conserved His and Asn residues [24] | DED catalytic motif (Asp, Glu, Asp) [25] [26] |
| Metal Cofactor Dependence | Mg²⁺ dependent [22] | Mg²⁺ dependent [22] [26] |
| Key Structural Insight | Undergoes major conformational activation upon target binding [22] | Functions as a dimer; achieves sequence specificity through dynamic DNA probing [26] |
Experimental Characterization of Cleavage Mechanisms
In Vitro Cleavage Assays for Mechanism Determination
The distinct cleavage mechanisms of the HNH and RuvC domains were elucidated through carefully designed in vitro cleavage assays.
Protocol for In Vitro DNA Cleavage Assay (adapted from [22])
- Protein Purification: Express and purify recombinant Cas9 protein (e.g., from Streptococcus thermophilus LMG18311) using affinity and size-exclusion chromatography.
- RNA Preparation: Generate single guide RNA (sgRNA) or crRNA/tracrRNA complexes via in vitro transcription with T7 RNA polymerase and subsequent gel purification.
- Assay Setup: Reconstitute the cleavage reaction by incubating purified Cas9 (e.g., 5 µM) with the guide RNA and a target DNA plasmid containing the protospacer and a cognate PAM sequence in an appropriate reaction buffer.
- Mutagenesis Analysis: To assign cleavage activity to specific domains, generate and purify catalytic dead mutants (e.g., HNH mutant H825A or RuvC mutant D15A) and repeat the assay. The loss of specific strand cleavage implicates the mutated domain.
- Product Analysis: Resolve the reaction products using agarose gel electrophoresis. Successful cleavage of the target plasmid will generate two smaller, quantifiable DNA fragments.
Structural Analysis of RuvC's Sequence Specificity
The sequence preference of the ancestral RuvC resolvase (for the consensus 5'-A/TTT↓G/C-3') provides a model for understanding nuclease specificity. Biochemical and structural studies, including X-ray crystallography and Molecular Dynamics (MD) simulations, reveal that RuvC does not make direct base-specific contacts. Instead, it achieves specificity through a dynamic probing mechanism [26].
Key Experimental Workflow for RuvC HJ Resolution [26]
- Crystallization: Co-crystallize RuvC with a synthetic Holliday junction (HJ) DNA substrate.
- Data Collection & Structure Solution: Collect X-ray diffraction data and solve the structure of the RuvC-HJ complex.
- MD Simulations: Perform microsecond-scale MD simulations on the solved structure, replacing non-cognate DNA sequences with cognate ones to observe conformational changes.
- Biochemical Validation: Test predictions from the MD simulations (e.g., the role of specific residues like Arg76 in base flipping) using site-directed mutagenesis and in vitro cleavage assays with cognate versus non-cognate HJ substrates.
This combined approach revealed that RuvC induces strain at the HJ exchange point. For cognate sequences, the complex can access rare, high-energy states where a specific thymidine base flips out, allowing the scissile phosphate to move into the catalytic site. This conformational change is less feasible for non-cognate sequences, providing a kinetic barrier that ensures cleavage specificity [26].
Diagram 1: HNH and RuvC-like domain cleavage mechanisms leading to DSB formation.
The Scientist's Toolkit: Essential Reagents for Nuclease Mechanism Studies
Table 2: Key Research Reagents for Studying HNH and RuvC Mechanisms
| Reagent / Tool | Function / Purpose | Example & Notes |
|---|---|---|
| Catalytic Dead Mutants | Assigns cleavage activity to a specific nuclease domain by ablating its function. | HNH mutant (e.g., H840A in SpCas9), RuvC mutant (e.g., D10A in SpCas9). Used in in vitro cleavage assays [22]. |
| Recombinant Cas9 Orthologs | Comparative studies of cleavage mechanics and PAM requirements across different systems. | Streptococcus thermophilus LMG18311 Cas9, with distinct PAM specificity [22]. |
| Guide RNA (sgRNA/crRNA) | Directs Cas9 to a specific DNA target sequence for cleavage. | Designed as a ~20 nt sequence complementary to the target; can be produced by in vitro transcription [22] [23]. |
| Synthetic Holliday Junctions (HJs) | Substrate for studying the mechanism and specificity of ancestral RuvC resolvase. | Synthetic four-way DNA junctions with cognate (5'-A/TTT↓G/C-3') or non-cognate sequences for in vitro assays [26]. |
| Divalent Metal Cofactors | Essential catalytic cofactor for both HNH and RuvC-like nuclease activities. | Mg²⁺ is the primary physiological cofactor; required in reaction buffers for cleavage assays [22] [26]. |
Implications for Therapeutic Development and Research
The detailed mechanistic understanding of HNH and RuvC domains directly fuels advances in CRISPR-based therapeutic development. The ability to create nickase variants (e.g., Cas9n, where one domain is inactivated) enables single-strand breaks for more precise editing with reduced off-target effects [23] [27]. Furthermore, this knowledge underpins the engineering of high-fidelity Cas9 variants and novel editors like prime editors, which bypass DSB formation altogether, thereby enhancing safety profiles for clinical applications [28] [29].
In drug discovery and functional genomics, CRISPR-Cas9 is instrumental for high-throughput screens to identify and validate new drug targets [30] [23] [29]. The precision of the dual nuclease system allows for the creation of more accurate disease models and the development of cell-based therapies, such as engineered CAR-T cells where specific genes are knocked out to enhance anti-tumor activity [23] [31]. A novel therapeutic approach involves weaponizing Cas9 to induce lethal DSBs specifically in aberrant cells (e.g., cancer cells or those harboring silent viral DNA) based on their unique DNA sequences, a strategy that is independent of gene expression or function [27].
Diagram 2: From mechanistic insight to application in research and therapy.
Structural Conformational Changes Driving the DNA Cleavage Process
The CRISPR-Cas9 system has revolutionized genome engineering by providing unprecedented precision in manipulating DNA sequences. At the heart of this technology lies the Cas9 endonuclease, which undergoes a series of sophisticated structural rearrangements to execute DNA cleavage. These conformational changes represent a critical regulatory mechanism that ensures the specificity of DNA targeting and cleavage, forming the foundation for reliable genome editing applications in therapeutic development [28].
This technical guide examines the structural basis of Cas9 activation, focusing on the dynamic transitions that enable the formation of DNA double-strand breaks (DSBs). Within the context of CRISPR-Cas9 mechanism research, understanding these conformational changes is paramount for optimizing editing efficiency and specificity, particularly for drug development applications where off-target effects present significant safety concerns [32]. The precise molecular choreography between Cas9's domains serves as a final proofreading step before irreversible DNA cleavage occurs, making it a fundamental process for researchers to comprehend [33].
Structural Fundamentals of the Cas9 Enzyme
The Cas9 enzyme possesses a bilobed architecture consisting of two primary structural elements: the recognition (REC) lobe and the nuclease (NUC) lobe. The REC lobe, composed of REC1, REC2, and REC3 domains, is responsible for nucleic acid binding and recognition. The NUC lobe contains the HNH and RuvC nuclease domains that perform the DNA cleavage activity [34] [32].
Cas9 operates as an RNA-guided DNA endonuclease that requires a single-guide RNA (sgRNA) for sequence-specific targeting. The sgRNA directs Cas9 to complementary DNA sequences adjacent to a protospacer adjacent motif (PAM), which is essential for initial DNA recognition [32]. Upon PAM binding, Cas9 initiates DNA unwinding, allowing the guide RNA to form a heteroduplex with the target DNA strand (TS), while displacing the non-target strand (NTS) [34].
The catalytic heart of Cas9 resides in its two nuclease domains: HNH cleaves the TS complementary to the sgRNA, while RuvC cleaves the NTS. These domains are spatially separated in the inactive state but undergo substantial repositioning to achieve catalytic competence [35]. The HNH domain exhibits remarkable conformational flexibility, sampling multiple states before adopting the active configuration necessary for DNA cleavage [36].
Table 1: Key Domains and Structural Elements of Cas9
| Domain/Element | Function | Structural Features |
|---|---|---|
| REC Lobe | Guide RNA and target DNA binding | Comprises REC1, REC2, REC3 domains; facilitates nucleic acid recognition |
| HNH Domain | Cleaves target DNA strand | Exhibits high conformational flexibility; contains catalytic residue H840 |
| RuvC Domain | Cleaves non-target DNA strand | Maintains relatively stable position; contains catalytic residue D10 |
| PAM Interface | Initial DNA recognition | Recognizes NGG sequence; allosterically activates nuclease domains |
| L1/L2 Linkers | Signal transduction | Connects structural elements; mediates allosteric communication |
Conformational States During DNA Cleavage Activation
Sequential Activation of the HNH Domain
The HNH domain undergoes a precisely orchestrated conformational transition to activate DNA cleavage. Structural studies have revealed at least three distinct states during this process. In the HNH-state 1 (inactive conformation), the HNH active site is positioned more than 32 Å from the DNA cleavage site, rendering it catalytically incompetent. In the intermediate HNH-state 2, the domain moves closer, but the catalytic site remains approximately 19 Å from the scissile phosphorus. Finally, in HNH-state 3 (active conformation), the HNH domain rotates approximately 170° around a central axis, bringing its active site to the optimal position for DNA cleavage [35].
This transition involves substantial structural rearrangements beyond simple translation. The linker region L2 (residues 906-923) undergoes a helix-to-loop conformational change that facilitates the dramatic reorientation of HNH. In the active state, the HNH domain establishes new contacts with the REC1 and PI domains through segments comprising residues 861-864, 872-876, and 903-906 [35]. These interactions stabilize the domain in its catalytically competent configuration.
Allosteric Coordination Between Nuclease Domains
The cleavage of both DNA strands is tightly coordinated through allosteric communication between the HNH and RuvC domains. The HNH domain serves as an allosteric regulator of RuvC activity—only when HNH adopts its active conformation does RuvC become fully capable of cleaving the NTS [33]. This mechanism ensures concerted firing of both nuclease domains, preventing single-strand nicks unless both domains are properly positioned.
Molecular dynamics simulations have revealed that the L1 and L2 loops function as critical "signal transducers" in this allosteric network [32]. PAM binding initiates a population shift that propagates through these structural elements, inducing highly coupled motions of HNH and RuvC. This allosteric cross-talk creates a proofreading mechanism that verifies correct target recognition before permitting irreversible DNA cleavage [32].
Table 2: Quantitative Parameters of HNH Conformational States
| HNH State | Distance from Cleavage Site | Domain Rotation | Key Structural Features |
|---|---|---|---|
| State 1 (Inactive) | >32 Å | Reference state | Crystallographic state; HNH distal from DNA |
| State 2 (Intermediate) | ~19 Å | Partial rotation | Closer approach; not fully activated |
| State 3 (Active) | Catalytic distance | ~170° rotation | Contacts REC1 and PI domains; L2 linker in loop conformation |
Experimental Characterization of Conformational Dynamics
Cryo-Electron Microscopy Approaches
Cryo-EM has been instrumental in visualizing Cas9 conformational states at near-atomic resolution. A landmark 5.2 Å resolution cryo-EM structure of the Cas9-sgRNA-DNA ternary complex revealed the HNH domain in its active conformation (State 3), providing the first structural evidence of its repositioning for catalysis [35].
Experimental Protocol: Cryo-EM Structure Determination
- Complex Preparation: Incubate Streptococcus pyogenes Cas9 (SpCas9) with nuclease activity-dead mutations (D10A, H840A) with a 55-bp target DNA and corresponding 98-nt sgRNA to form a stable ternary complex
- Vitrification: Rapidly freeze the complex on cryo-EM grids to preserve native structure
- Data Collection: Acquire multiple micrographs using cryo-electron microscope
- 2D Classification: Identify and group similar particle images
- 3D Reconstruction: Apply single-particle analysis to generate initial density map
- Refinement: Iteratively refine the model against the density map to achieve final resolution [35]
This approach successfully captured the PAM-proximal region in a stable, base-paired form, while the PAM-distal end appeared more flexible, suggesting dynamic behavior during R-loop formation [35].
Single-Molecule FRET Methodologies
Single-molecule FRET (smFRET) has provided unprecedented insights into the real-time dynamics of Cas9 conformational changes. By site-specifically labeling Cas9 with donor and acceptor fluorophores, researchers have monitored domain movements under various binding conditions [33] [36].
Experimental Protocol: smFRET for HNH Conformational Monitoring
- Protein Engineering: Introduce cysteine residues at strategic positions (e.g., S355-S867 or S867-N1054) in a cysteine-free Cas9 background for specific dye labeling
- Fluorophore Conjugation: Label engineered cysteines with Cy3 (donor) and Cy5 (acceptor) maleimide dyes
- Complex Formation: Incubate labeled Cas9 with sgRNA and various DNA substrates (on-target, off-target, or mismatched)
- FRET Measurements: Monitor energy transfer efficiency between fluorophores using total internal reflection fluorescence (TIRF) microscopy
- Data Analysis: Calculate (ratio)A values (acceptor fluorescence via energy transfer normalized to direct excitation) to quantify conformational states [33]
This methodology revealed that the HNH domain samples an equilibrium between active and inactive conformations, with on-target DNA stabilizing the active state and off-target substrates favoring inactive conformations [33]. The measured FRET efficiencies directly correlated with DNA cleavage activities, establishing a quantitative relationship between HNH positioning and catalytic function.
Diagram 1: smFRET Experimental Workflow for Cas9 Conformational Analysis
Molecular Dynamics Simulations
Computational approaches have complemented experimental methods in characterizing Cas9 conformational dynamics. Molecular dynamics (MD) simulations, particularly Gaussian accelerated MD (GaMD), have enabled the exploration of large-scale transitions occurring on microsecond to millisecond timescales [32].
Computational Protocol: Enhanced Sampling MD Simulations
- System Preparation: Construct atomic models of Cas9 in various ligand-bound states using available crystal structures
- Solvation and Ionization: Embed the protein in explicit solvent with physiological ion concentrations
- Equilibration: Gradually relax the system through restrained and unrestrained MD simulations
- Enhanced Sampling: Apply GaMD methods to overcome energy barriers and observe rare transitions
- Pathway Analysis: Identify intermediate states and conformational pathways using dimensionality reduction techniques
- Validation: Compare simulated conformations with experimental structures and FRET data [32]
These simulations have predicted the existence of an active HNH conformation that was later confirmed experimentally, demonstrating the predictive power of modern computational approaches [32]. MD simulations have also revealed large-scale movements of the REC lobe (Rec2 domain translation of ~8-10 Å, Rec3 translation of ~5 Å) that accommodate HNH docking at the catalytic site [32].
The Scientist's Toolkit: Key Research Reagents and Methodologies
Table 3: Essential Research Reagents for Studying Cas9 Conformational Changes
| Reagent/Method | Specific Application | Key Function | Technical Considerations |
|---|---|---|---|
| High-Fidelity Cas9 Variants | Specificity studies | Reduced off-target cleavage; point mutations in REC3 or RuvC domains | Examples: eCas9, HypaCas9, evoCas9; maintain on-target efficiency |
| Fluorophore-Labeled Cas9 | smFRET experiments | Site-specific labeling for distance measurements | Cysteine-free background required; confirm functional activity post-labeling |
| Cryo-EM Grids | Structural studies | Vitrification of ternary complexes | Optimize freezing conditions to preserve native state |
| Modified DNA Substrates | Cleavage kinetics | On-target, off-target, and mismatched sequences | Design includes PAM and variable complementarity regions |
| MD Simulation Software | Computational modeling | All-atom dynamics with enhanced sampling | Requires significant computational resources; validate with experimental data |
Implications for Therapeutic Genome Editing
Understanding Cas9 conformational dynamics has direct implications for therapeutic genome editing applications. The proofreading mechanism mediated by HNH dynamics serves as a natural barrier against off-target effects, a major concern in clinical applications [33] [36]. Engineering efforts have leveraged this knowledge to develop high-fidelity Cas9 variants with improved specificity profiles.
The allosteric communication between Cas9 domains presents opportunities for therapeutic intervention. Small molecules that modulate these allosteric pathways could potentially fine-tune Cas9 activity, offering additional control over genome editing outcomes [32]. Additionally, the conformational checkpoint mechanism could be exploited to develop novel genome editing platforms with built-in safety features.
Recent research has also demonstrated the potential for weaponizing CRISPR/Cas9 to selectively eliminate aberrant cells by targeting genomic sequences unique to cancer cells or viral pathogens [27]. This approach leverages the precise DNA recognition and cleavage capabilities of Cas9, while understanding conformational dynamics ensures selective targeting without affecting healthy cells.
Diagram 2: Cas9 Conformational Activation Pathway for DNA Cleavage
The structural conformational changes driving DNA cleavage in CRISPR-Cas9 represent a sophisticated molecular mechanism that balances catalytic efficiency with target specificity. The dynamic transitions of the HNH domain, coupled with allosteric regulation of RuvC activity, create a proofreading system that verifies correct target recognition before permitting DNA cleavage. This understanding has enabled the development of improved genome editing tools with enhanced specificity profiles.
For researchers and drug development professionals, mastering these conformational dynamics is essential for advancing CRISPR-based therapeutic applications. The experimental and computational methodologies reviewed here provide a toolkit for investigating these processes, while engineered Cas9 variants offer improved platforms for precise genome manipulation. As structural biology techniques continue to evolve, particularly in cryo-EM and single-molecule imaging, our understanding of these fundamental processes will further refine the safety and efficacy of genome editing technologies for clinical applications.
From Mechanism to Medicine: Therapeutic Applications of CRISPR-Induced DSBs
The CRISPR-Cas9 system has revolutionized genetic research by providing unprecedented precision in genome editing. However, a critical understanding often overlooked is that the CRISPR-Cas9 machinery itself does not perform the genetic modification but rather serves as "molecular scissors" that create a targeted double-strand break (DSB) in the DNA [37] [38]. The actual genetic editing occurs through the cell's endogenous DNA damage repair (DDR) pathways, which are activated to resolve this break [38]. Two principal pathways compete to repair these breaks: error-prone Non-Homologous End Joining (NHEJ) and high-fidelity Homology-Directed Repair (HDR) [37] [28]. The fundamental choice between these pathways determines the editing outcome, making their manipulation essential for achieving predictable genetic modifications in CRISPR-based experiments. Within the broader context of DSB formation research, understanding and controlling these endogenous cellular processes is what transforms a simple DNA cut into a powerful tool for genetic engineering, with profound implications for basic research and therapeutic development.
DNA Repair Mechanism Fundamentals
Non-Homologous End Joining (NHEJ): The Rapid Response Mechanism
Non-Homologous End Joining is the cell's primary, fast-acting mechanism for repairing DSBs. This pathway functions throughout the cell cycle and operates by directly ligating the two broken ends of the DNA double helix without requiring a homologous template [37] [38]. Its key characteristic is its error-prone nature; the rejoining process often results in small insertions or deletions (INDELs) at the repair site [38]. These INDELs typically range from 1 to 10 base pairs and can disrupt gene function by causing frameshift mutations, leading to premature stop codons and effectively knocking out the gene [38]. The distinguishing features of NHEJ are its speed, as it is the cell's first line of defense against DSBs; its template independence, functioning without a homologous DNA template; and its high efficiency across all phases of the cell cycle [38]. While NHEJ is ideal for gene knockouts, it can also be co-opted for gene knock-in strategies with appropriately designed donor templates, though with less precision than HDR-based approaches [37].
Homology-Directed Repair (HDR): The Precision Repair Pathway
Homology-Directed Repair represents a high-fidelity alternative to NHEJ. Unlike the template-independent NHEJ pathway, HDR requires a homologous DNA template—such as a sister chromatid, a donor plasmid, or a single-stranded oligodeoxynucleotide (ssODN)—to accurately repair the DSB [37] [38]. This template-dependent mechanism allows for precise genetic modifications, including nucleotide substitutions, gene corrections, or the insertion of larger DNA fragments such as fluorescent protein tags [37]. However, HDR's defining features also present practical limitations. Its precision comes at the cost of lower efficiency compared to NHEJ, and it is cell cycle-dependent, occurring primarily during the S and G2 phases when homologous templates are available through DNA replication [38]. This pathway is consequently less efficient in non-dividing or slowly dividing cells, such as neurons or cardiomyocytes [39]. For researchers aiming to perform precise gene knock-ins, point mutations, or gene corrections, HDR is the indispensable pathway, despite requiring additional experimental strategies to enhance its efficiency.
Table 1: Comparative Analysis of NHEJ and HDR Pathways
| Feature | Non-Homologous End Joining (NHEJ) | Homology-Directed Repair (HDR) |
|---|---|---|
| Template Required | No | Yes (donor DNA with homology arms) |
| Primary Outcome | Small insertions/deletions (INDELs) | Precise sequence insertion/correction |
| Efficiency | High | Low to moderate |
| Cell Cycle Dependence | Active throughout all phases | Restricted to S and G2 phases |
| Kinetics | Fast (hours) | Slow (hours to days) |
| Key Applications | Gene knockouts, gene disruption | Gene knock-ins, precise point mutations, gene correction |
| Major Advantage | Highly efficient in most cell types | High precision and accuracy |
| Major Limitation | Error-prone, introduces random INDELs | Low efficiency, requires donor design |
Experimental Strategies and Methodologies
Designing Experiments for NHEJ-Mediated Gene Knockouts
To successfully implement NHEJ for gene knockout studies, researchers must follow a structured experimental approach. The essential components required are the Cas9 nuclease (delivered as protein, plasmid, or mRNA) and a single guide RNA (sgRNA) complexed with Cas9 [38]. The experimental workflow begins with the careful design of sgRNAs that target early exonic regions of the gene of interest to maximize the likelihood of generating frameshift mutations. After delivering these components into the target cells, the editing outcome must be validated. This is typically done by extracting genomic DNA and using PCR to amplify the target region, followed by sequencing analysis to detect the spectrum of INDELs introduced at the cut site [38]. The efficiency of gene disruption is often quantified using mismatch detection assays (e.g., T7E1 or Surveyor assays) or, more accurately, by high-throughput sequencing methods. A critical consideration is that traditional short-read sequencing may miss large, on-target deletions (kilobase- to megabase-scale) that have been recently identified as a common outcome of CRISPR editing [14]. These large structural variations (SVs) can have profound functional consequences but are often undetected by standard amplification and sequencing approaches if primer binding sites are lost [14].
Implementing HDR for Precise Genetic Modifications
The implementation of HDR requires more complex experimental design compared to NHEJ-based approaches. In addition to the Cas9 nuclease and sgRNA, HDR experiments necessitate the design and delivery of a donor DNA template containing the desired modification flanked by homology arms that match the sequences surrounding the cut site [37] [38]. The length of these homology arms varies depending on the application: for ssODN templates used to introduce point mutations or small insertions, arms of 30-60 nucleotides are typically sufficient, while for plasmid-based donors for larger insertions, arms of 500-1000 nucleotides are common. To maximize HDR efficiency, researchers often employ strategic interventions to shift the cellular repair balance away from the dominant NHEJ pathway. These include cell cycle synchronization to enrich for cells in S/G2 phase where HDR is active [38], and the use of small molecule inhibitors targeting key NHEJ proteins such as DNA-PKcs (e.g., NU7441, M3814) [14] [38]. However, recent studies have revealed that some enhancement strategies, particularly the use of DNA-PKcs inhibitors like AZD7648, can inadvertently increase the frequency of large-scale chromosomal aberrations and translocations [14]. This underscores the importance of comprehensive genotoxic profiling when developing therapeutic editing approaches.
Addressing Unique Challenges in Non-Dividing Cells
Recent research has highlighted significant differences in DNA repair dynamics between dividing and non-dividing cells, with important implications for therapeutic editing in postmitotic cells such as neurons and cardiomyocytes. A 2025 study comparing induced pluripotent stem cells (iPSCs) and iPSC-derived neurons revealed that Cas9-induced indels accumulate over a significantly longer time course in neurons—continuing to increase for up to two weeks post-transduction, compared to a few days in dividing cells [39]. Furthermore, neurons exhibit a different distribution of repair outcomes, with a strong bias toward small NHEJ-mediated indels and a suppression of the larger deletions typically associated with microhomology-mediated end joining (MMEJ) that are more common in dividing cells [39]. These findings necessitate adapted experimental timelines and analytical approaches when working with clinically relevant non-dividing cells.
Advanced Technical Considerations and Risk Mitigation
The Scientist's Toolkit: Essential Reagents for CRISPR Repair Studies
Table 2: Key Research Reagents for Controlling and Assessing DNA Repair Outcomes
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| NHEJ Inhibitors | DNA-PKcs inhibitors (NU7441, M3814), 53BP1 inhibitors | Enhance HDR efficiency by suppressing competing NHEJ pathway [14] [38] |
| HDR Donor Templates | Single-stranded ODNs (ssODNs), double-stranded DNA plasmids with homology arms | Provide template for precise repair; design depends on edit size [38] |
| Cell Synchronization Agents | Aphidicolin, Thymidine, Nocodazole | Enrich cell populations in S/G2 phase where HDR is active [38] |
| Delivery Vehicles | Virus-like particles (VLPs), Electroporation, Chemical transfection | Enable efficient RNP delivery, especially in challenging cells like neurons [39] |
| Analysis Tools | Amplicon sequencing, CAST-Seq, LAM-HTGTS | Detect editing outcomes and structural variations [14] |
Emerging Risks: Structural Variations and Genomic Instability
Beyond the well-documented concerns about off-target effects, recent studies have revealed more pressing challenges associated with CRISPR editing, particularly the formation of large structural variations (SVs) including chromosomal translocations and megabase-scale deletions [14]. These SVs represent substantial safety concerns for clinical translation and are often underestimated in standard editing assessments. The risk of such events appears particularly elevated in cells treated with DNA-PKcs inhibitors to enhance HDR, where surveys have shown not only a qualitative rise in the number of translocation sites but also an alarming thousand-fold increase in the frequency of these SVs [14]. Additionally, techniques that rely on short-read sequencing can dramatically overestimate HDR efficiency while concurrently underestimating INDEL frequencies when large deletions remove primer binding sites, rendering these events 'invisible' to standard analysis [14]. These findings highlight the critical need for comprehensive SV screening using specialized methods like CAST-Seq or LAM-HTGTS in therapeutic editing applications [14].
Visualization of Repair Pathways and Experimental Workflows
CRISPR-Cas9 DNA Repair Pathway Logic
Experimental Workflow for CRISPR Repair Studies
The strategic harnessing of endogenous DNA repair pathways represents the cornerstone of successful CRISPR-Cas9 genome editing. The intentional selection between NHEJ for gene knockouts and HDR for precise modifications enables researchers to address diverse biological questions and therapeutic needs. However, the growing understanding of cell-type-specific repair dynamics, particularly in non-dividing cells, and the emerging risks of structural variations necessitate increasingly sophisticated experimental approaches. As the field progresses, the integration of advanced delivery systems, refined small-molecule interventions, and comprehensive genomic safety profiling will be essential for realizing the full potential of CRISPR-based therapies while mitigating unintended consequences. The continued elucidation of DSB repair mechanisms in diverse cell types will undoubtedly yield new strategies for achieving unprecedented control over genome editing outcomes.
The CRISPR-Cas9 system, derived from a natural bacterial defense mechanism, has revolutionized biological research and therapeutic development by enabling precise, targeted modifications to the genome [40]. This system functions as a programmable genome editing tool comprised of two key components: a Cas9 nuclease that creates double-strand breaks (DSBs) in DNA and a guide RNA (gRNA) that directs Cas9 to a specific genomic locus [41] [40]. The therapeutic application of this technology represents a paradigm shift in medicine, particularly for monogenic disorders.
Sickle Cell Disease (SCD) and Transfusion-Dependent Beta-Thalassemia (TDT) are hereditary hemoglobinopathies that emerged as ideal candidates for CRISPR-based therapies [40]. SCD is caused by a point mutation in the β-globin gene (HBB), leading to the production of sickle hemoglobin (HbS) that polymerizes under low oxygen conditions, resulting in sickled red blood cells that cause vaso-occlusive crises and organ damage [41]. TDT is characterized by reduced or absent synthesis of β-globin chains, causing severe anemia and lifelong dependence on blood transfusions [40]. Both diseases affect hemoglobin function through different mechanisms but share a common therapeutic target: the reactivation of fetal hemoglobin (HbF), which is naturally present during fetal development but silenced after birth [40].
The FDA approval of Casgevy (exagamglogene autotemcel, or exa-cel) in December 2023 marked a historic milestone as the first CRISPR-based medicine approved in the United States for treating both SCD and TDT [42] [43]. This approval was swiftly followed by the authorization of Lyfgenia (lovotibeglogene autotemcel, or lovo-cel), a lentiviral vector-based gene therapy, establishing a new class of genetically modified cell-based therapies for these devastating disorders [42].
Mechanism of Action: From Double-Strand Breaks to Therapeutic Effect
CRISPR-Cas9-Mediated Genome Editing Strategy
Casgevy employs a sophisticated mechanism that leverages the cellular response to CRISPR-induced double-strand breaks to achieve therapeutic benefit. The process involves ex vivo editing of autologous hematopoietic stem cells (HSCs) harvested from the patient [42] [43].
The therapeutic strategy focuses on the BCL11A gene, which encodes a transcriptional repressor that silences fetal hemoglobin production after birth [43]. Casgevy uses a CRISPR-Cas9 complex programmed with a guide RNA that directs the Cas9 nuclease to create a precise double-strand break in the BCL11A gene [42] [40]. This break disrupts the gene's function, thereby releasing the repression on fetal hemoglobin production [43].
The resulting edited HSCs, when reinfused into the patient, engraft in the bone marrow and give rise to red blood cells that produce high levels of fetal hemoglobin [40]. HbF functions as an effective anti-sickling agent in SCD by interfering with the polymerization of HbS [40], and it compensates for the hemoglobin deficiency in TDT, reducing or eliminating the need for transfusions [40].
Table: Core Components of the CRISPR-Cas9 Therapeutic System in Casgevy
| Component | Type/Role | Therapeutic Function |
|---|---|---|
| Cas9 Nuclease | DNA endonuclease | Creates double-strand breaks at targeted genomic locations [41] |
| Guide RNA (gRNA) | Targeting RNA molecule | Directs Cas9 to specific sequence in BCL11A gene [41] |
| BCL11A Target | Erythroid-specific transcription factor | Gene encoding repressor of fetal hemoglobin production [43] |
| Fetal Hemoglobin (HbF) | Therapeutic protein product | Developmentally regulated hemoglobin with anti-sickling properties [40] |
Lyfgenia: An Alternative Gene Therapy Approach
Lyfgenia employs a different mechanism, utilizing a lentiviral vector to deliver a functional gene encoding HbAT87Q, a modified hemoglobin molecule designed to resemble normal adult hemoglobin but with anti-sickling properties [42]. Unlike Casgevy's gene disruption approach, Lyfgenia represents a gene addition strategy, where the lentiviral vector inserts the therapeutic gene into the genome of patient HSCs [42]. The FDA included a black box warning for Lyfgenia regarding the risk of hematologic malignancy, reflecting concerns about viral vector-mediated insertional mutagenesis [42].
The diagram below illustrates the fundamental mechanism of Casgevy compared to conventional gene therapy:
Clinical Trial Data and Efficacy Outcomes
Casgevy Clinical Trial Results
The safety and efficacy of Casgevy were established in ongoing single-arm, multi-center trials. For SCD, the primary efficacy outcome measured was freedom from severe vaso-occlusive crises (VOCs) for at least 12 consecutive months during the 24-month follow-up period [42]. Of the 31 patients with sufficient follow-up time to be evaluable, 29 (93.5%) achieved this outcome [42]. All treated patients achieved successful engraftment with no instances of graft failure or rejection [42].
For TDT, the clinical trials demonstrated that 25 out of 27 patients treated with Casgevy were no longer transfusion-dependent following treatment, with some patients maintaining transfusion independence for over three years [40]. The remaining two patients showed dramatic reductions in transfusion frequency of 80% and 96%, respectively [40].
Lyfgenia Clinical Trial Results
Lyfgenia was evaluated in a single-arm, 24-month multicenter study in patients with SCD and a history of vaso-occlusive events [42]. Effectiveness was based on complete resolution of VOEs (VOE-CR) between 6 and 18 months after infusion. The results showed that 28 (88%) of 32 patients achieved VOE-CR during this time period [42]. Additional data from bluebird bio submitted to the FDA showed lovo-cel was effective in 36 people who were followed for a median of 32 months [43].
Table: Comparative Clinical Outcomes of FDA-Approved CRISPR/Gene Therapies
| Parameter | Casgevy (exa-cel) | Lyfgenia (lovo-cel) |
|---|---|---|
| Technology Platform | CRISPR-Cas9 genome editing [42] | Lentiviral vector gene addition [42] |
| Molecular Target | BCL11A gene [43] | β-globin gene with anti-sickling modification [42] |
| Therapeutic Mechanism | Fetal hemoglobin (HbF) reactivation [40] | Production of HbA^T87Q anti-sickling hemoglobin [42] |
| SCD Efficacy (VOCs) | 93.5% free of severe VOCs for ≥12 months [42] | 88% with complete VOE resolution (6-18 months) [42] |
| TDT Efficacy | 93% transfusion independence [40] | Not approved for TDT |
| Notable Safety Findings | No graft failure/rejection [42] | Black box warning for hematologic malignancy [42] |
Quality of Life Outcomes
Recent studies have documented significant improvements in quality of life following treatment with exa-cel (Casgevy). Research published in Blood Advances in 2025 demonstrated robust and sustained improvements in overall quality of life, including physical, social/family, functional, and emotional well-being [44]. These improvements were observed starting as early as six months following exa-cel infusion and were maintained through long-term follow-up (median 33.6 months for SCD and 38.4 months for TDT) [44].
For SCD patients, results from the ASCQ-Me quality of life scale showed the greatest non-pain improvements in social impact (+16.5), emotional impact (+8.5), and sleep impact (+5.7) [44]. Adolescent patients showed remarkable improvements in school functioning (+45), social functioning (+18.3), and emotional functioning (+16.7) after infusion [44].
Experimental Protocols and Methodologies
Hematopoietic Stem Cell Collection and Processing
The therapeutic protocol begins with the collection of autologous CD34+ hematopoietic stem and progenitor cells via apheresis following mobilization with granulocyte colony-stimulating factor (G-CSF) and plerixafor [45] [42]. The collected cells are then transported to a specialized manufacturing facility where they undergo CRISPR-Cas9 genome editing using electroporation to deliver the ribonucleoprotein (RNP) complex composed of Cas9 nuclease and guide RNA [42].
CRISPR Genome Editing Workflow
The precise experimental protocol for Casgevy manufacturing involves:
- Cell Preparation: Isolated CD34+ HSPCs are cultured in serum-free media supplemented with cytokines (SCF, TPO, FLT3-L) to maintain viability and stemness [45].
- Electroporation: Cells are electroporated with precomplexed CRISPR-Cas9 RNP targeting the BCL11A gene. Optimization studies determined that VSVG/BRL-co-pseudotyped FMLV virus-like particles (VLPs) achieve up to 97% transduction efficiency in human cells [39].
- Quality Control: Edited cells undergo rigorous testing including Sanger sequencing and next-generation sequencing (NGS) to verify on-target editing efficiency and detect potential off-target effects [43].
- Expansion and Formulation: Successfully edited cells are expanded ex vivo before cryopreservation in infusion-ready media [42].
Patient Conditioning and Reinfusion
Prior to edited cell reinfusion, patients undergo myeloablative conditioning with busulfan to create marrow niche space for the engineered cells [42] [40]. The cryopreserved Casgevy product is then thawed and administered via intravenous infusion [42]. Patients are monitored closely for engraftment, typically evidenced by neutrophil and platelet recovery within several weeks post-infusion [45].
The following diagram illustrates the comprehensive therapeutic workflow:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table: Key Research Reagent Solutions for CRISPR Therapeutic Development
| Reagent/Material | Specific Example | Research Function |
|---|---|---|
| CRISPR Nucleases | Cas9, Cas12a [45] [40] | Programmable DNA endonucleases for creating targeted double-strand breaks |
| Guide RNA (gRNA) | BCL11A-targeting sgRNA [43] | RNA component that provides targeting specificity through complementary base pairing |
| Delivery Systems | Virus-like particles (VLPs) [39], Electroporation | Vehicles for intracellular delivery of CRISPR components |
| Stem Cell Media | Serum-free media with SCF, TPO, FLT3-L [45] | Specialized formulations for ex vivo hematopoietic stem cell maintenance and expansion |
| Editing Assessment | Next-generation sequencing (NGS) [43], Sanger sequencing | Analytical tools for quantifying on-target editing and detecting off-target effects |
| Cell Selection | CD34+ magnetic bead selection [45] | Isolation of hematopoietic stem and progenitor cells from apheresis product |
| Animal Models | Humanized mouse models [39] | Preclinical systems for evaluating engraftment and safety of edited cells |
Emerging Research and Future Directions
Advancements in DNA Repair Understanding
Recent research has revealed crucial insights into how DNA repair mechanisms differ between cell types, significantly impacting CRISPR editing outcomes. A 2025 study published in Nature Communications demonstrated that postmitotic cells such as neurons and cardiomyocytes repair Cas9-induced DNA damage differently than dividing cells [39]. These nondividing cells take longer to fully resolve DNA damage and upregulate non-canonical DNA repair factors in the process [39]. While this particular study focused on neuronal cells, its implications extend to hematopoietic stem cells, as understanding these differential repair pathways enables better control over editing outcomes.
Next-Generation CRISPR Therapies in Development
Several next-generation CRISPR therapies for hemoglobinopathies are advancing through clinical development:
- Reni-cel (EDIT-301): An investigational therapy using CRISPR-Cas12a to edit the gamma globin gene promoters to upregulate fetal hemoglobin [45]. Updated data from the phase 1/2/3 RUBY trial showed 27 of 28 patients were free of vaso-occlusive events post-infusion [45].
- BEAM-101: A base editing therapy that creates single-nucleotide changes in the HBG1/2 gene promoters to inhibit BCL11A binding without creating double-strand breaks [45]. Recent trial data showed robust increases in fetal hemoglobin and reductions in sickle hemoglobin [45].
Personalized CRISPR Applications
A landmark case reported in May 2025 demonstrated the potential for personalized CRISPR therapeutics beyond SCD and TDT [46]. Researchers at Children's Hospital of Philadelphia developed a bespoke base editing therapy for an infant with carbamoyl phosphate synthetase 1 (CPS1) deficiency, a rare metabolic disorder [46]. The therapy was designed, manufactured, and administered within six months, with the patient showing improvement in symptoms and decreased dependence on medications after three doses [46]. This case establishes a precedent for on-demand gene editing therapies for rare genetic diseases.
The approval of Casgevy and Lyfgenia represents a transformative achievement in molecular medicine, demonstrating the therapeutic potential of CRISPR-Cas9 technology for addressing monogenic diseases at their genetic roots. These therapies exemplify the successful translation of basic research on CRISPR mechanisms and DNA repair pathways into clinically meaningful treatments that profoundly impact patients' lives.
The continued evolution of CRISPR technology—including base editing, prime editing, and personalized approaches—promises to expand the therapeutic landscape for genetic disorders. However, challenges remain in optimizing delivery, ensuring long-term safety, and improving accessibility of these complex therapies. As research advances our understanding of DNA repair mechanisms and editing specificity, the next generation of CRISPR therapies will likely offer even greater precision and efficacy for patients with SCD, TDT, and other genetic disorders.
The clinical success of CRISPR-Cas9 gene editing is fundamentally constrained by a central challenge: the safe and efficient delivery of its macromolecular components—the Cas nuclease and guide RNA (gRNA)—to target cells in living organisms. The CRISPR-Cas9 system induces double-strand breaks (DSBs) at precise genomic locations, activating cellular DNA repair pathways that can be harnessed for therapeutic gene correction or disruption. However, the inability of these components to passively enter cells necessitates a robust delivery vehicle. Lipid nanoparticles (LNPs) have emerged as a leading platform for systemic in vivo delivery, offering a non-viral vector that protects its cargo, facilitates cellular uptake, and can be engineered for tissue-specific targeting. This whitepaper examines the latest breakthroughs in LNP design that are enhancing the efficiency, specificity, and safety of systemic CRISPR-Cas9 delivery, thereby strengthening the foundation for its next-generation clinical applications.
Core LNP Architecture and Its Interaction with CRISPR-Cas9 Mechanisms
The functional efficacy of an LNP is dictated by its physicochemical properties—size, surface charge, and composition—which collectively influence its biodistribution, cellular internalization, and intracellular cargo release. Understanding this architecture is essential for designing LNPs that can successfully deliver the CRISPR machinery to the nucleus of target cells.
- Size and Surface Charge: The in vivo fate of LNPs is heavily influenced by their size and surface charge (zeta potential). Optimized particles, typically in the range of 50-100 nm, demonstrate favorable pharmacokinetics and tissue penetration profiles [47]. A positive surface charge enhances cellular uptake through electrostatic interactions with the negatively charged cell membrane.
- Key Lipid Components: A standard LNP formulation comprises several lipid types, each serving a distinct function, as detailed in the table below.
Table 1: Key Lipid Components and Their Functions in CRISPR-Cas9 LNP Formulations
| Lipid Component | Core Function | Impact on CRISPR Delivery |
|---|---|---|
| Ionizable Cationic Lipid | Encapsulates nucleic acid cargo; enables endosomal escape via protonation in acidic endosomes. | Critical for releasing Cas9-gRNA RNP complexes into the cytoplasm. New lipids like PL32 boost efficacy [48]. |
| Helper Lipid (e.g., DSPC) | Stabilizes the LNP bilayer structure. | Improves particle stability in systemic circulation, protecting CRISPR cargo from degradation [49]. |
| Cholesterol | Enhances membrane integrity and fluidity. | Stabilizes the LNP structure and facilitates cellular uptake [49]. |
| PEG-Lipid | Shields LNP surface, reduces aggregation, and modulates pharmacokinetics. | Prevents opsonization and rapid clearance, extending circulation half-life for improved target engagement [49] [50]. |
The journey of an LNP from systemic administration to nuclear gene editing follows a critical pathway. The diagram below illustrates this workflow, from formulation to functional genomic editing.
Recent Breakthroughs in LNP Formulations for Enhanced Systemic Delivery
Recent innovations have substantially improved the performance of LNPs, moving beyond standard formulations to overcome biological barriers and increase gene-editing efficiency.
Spherical Nucleic Acid (SNA) Architecture
A groundbreaking development from Northwestern University involves restructuring LNPs into spherical nucleic acids (LNP-SNAs). This architecture involves coating the LNP core with a dense, protective shell of DNA, which dramatically alters its cellular interactions [51]. In comparative studies, LNP-SNAs demonstrated a threefold increase in cellular uptake and a tripling of gene-editing efficiency across various human cell types, including stem cells and primary lymphocytes, compared to conventional LNPs. Furthermore, the system improved the success rate of precise homology-directed repair (HDR) by over 60%, a critical advance for therapeutic gene correction [51].
Targeted LNP (Ab-LNP) Delivery Strategies
A significant limitation of first-generation LNPs is their predominant accumulation in the liver. To redirect LNPs to extrahepatic tissues, researchers have developed antibody-targeted LNPs (Ab-LNPs). This strategy conjugates specific antibodies or antibody-like molecules to the LNP surface, enabling active targeting of cell-specific receptors [50]. The Weissman lab has pioneered several targeting approaches:
- Targeting the Lungs: Decorating LNPs with an anti-PECAM-1 antibody shifted biodistribution, resulting in a 200-fold increase in mRNA delivery and a 25-fold elevation of protein expression in mouse lungs compared to untargeted LNPs [50].
- Targeting T Cells: Using an anti-CD4 antibody, researchers achieved a 30-fold enhancement in reporter gene expression in CD4+ T cells, opening avenues for in vivo CAR-T cell engineering [50].
- Targeting Hematopoietic Stem Cells (HSCs): Anti-CD117 LNPs successfully delivered base-editing machinery to HSCs, nearly fully correcting the sickle cell disease mutation in vitro, and present a potential alternative to toxic conditioning regimens for bone marrow transplantation [50].
Mitigating Immunogenicity and Enhancing Efficiency
The inherent reactogenicity of ionizable lipids poses a challenge for therapeutic mRNA delivery, particularly in chronic inflammatory diseases. Recent work has led to the creation of a non-inflammatory LNP (NIF-LNP) by incorporating ursolic acid, a natural product, into a phosphoramide-derived lipid formulation [48]. This NIF-LNP exhibited a 40-fold enhancement in lung protein expression without causing significant inflammation. A genome-wide CRISPR screen identified that ursolic acid acts by activating the V-ATPase complex, promoting endosome acidification and trafficking without inducing immunogenicity [48].
Table 2: Quantitative Performance of Advanced LNP Platforms
| LNP Platform | Key Improvement | Quantitative Enhancement | Model System |
|---|---|---|---|
| LNP-SNA [51] | Cellular Uptake & Editing Efficiency | 3x higher uptake & 3x higher editing efficiency; >60% improvement in HDR | Human bone marrow stem cells, keratinocytes |
| Ab-LNP (Lung) [50] | Tissue-Specific Targeting | 200x increase in mRNA delivery to lungs | Mouse model |
| Ab-LNP (T Cell) [50] | Cell-Specific Targeting | 30x increase in gene expression in CD4+ T cells | In vitro human T cell culture |
| NIF-LNP [48] | Efficiency vs. Reactogenicity | 40x higher protein expression without inflammation | Mouse models of lung disease |
Experimental Protocols for Key LNP Applications
Protocol: In Vivo Gene Knockdown in the Liver
This protocol outlines a standard method for systemic delivery of CRISPR-Cas9 LNPs to induce gene knockdown in the liver, a common target for metabolic and genetic diseases.
- Step 1: LNP Formulation. Formulate LNPs using a microfluidic device. Mix an ionizable lipid (e.g., ALC-0315), DSPC, cholesterol, and DMG-PEG2000 at a molar ratio of 50:10:38.5:1.5 in ethanol (organic phase). The aqueous phase contains CRISPR-Cas9 mRNA and sgRNA complexed together or as a single RNP. Use a total flow rate of 500 µL/min and a flow rate ratio (aqueous:organic) of 3:1 to ensure homogeneous particle size [48] [52].
- Step 2: Characterization and Purification. Dialyze the formed LNPs against a buffer solution (e.g., PBS or HEPES) to remove residual ethanol. Characterize the final product for particle size (aiming for 70-100 nm), polydispersity index (PdI < 0.2), zeta potential, and encapsulation efficiency (>80%) using dynamic light scattering and Ribogreen assays [52].
- Step 3: Systemic Administration. Administer the LNP formulation to the animal model (e.g., mouse) via intravenous injection (e.g., tail vein). A standard dose for gene editing in mice ranges from 0.5 to 1.0 mg RNA per kg body weight [8] [48].
- Step 4: Efficacy and Safety Analysis. After 48-72 hours, analyze tissue samples. Assess editing efficiency by next-generation sequencing of the target locus from extracted genomic DNA. Evaluate potential off-target effects using methods like CAST-Seq or whole-genome sequencing. Monitor serum biomarkers for liver toxicity (e.g., ALT, AST) and pro-inflammatory cytokines to assess safety [14].
Protocol: Mitochondrial Genome Editing with RNP-MITO-Porter
The delivery of CRISPR to mitochondria presents a unique challenge due to the double mitochondrial membrane. The following protocol, adapted from [52], details a method for direct mitochondrial genome editing.
- Step 1: RNP Complex Formation. Incubate purified Cas9 protein with sgRNA designed to target a specific mitochondrial DNA (mtDNA) mutation (e.g., m.7778G>T in mouse mt-Atp8) at a molar ratio of 1:2 for 10-15 minutes at room temperature to form the RNP complex.
- Step 2: RNP Encapsulation. Prepare the MITO-Porter system using lipids DOPE, sphingomyelin, and stearylated-octaarginine (STR-R8). Load the pre-formed RNP into the MITO-Porter using a microfluidic device (e.g., iLiNP device) with an aqueous phase (HEPES buffer with RNP) and an organic phase (lipids in ethanol). The STR-R8 facilitates binding to the mitochondrial membrane and RNP encapsulation [52].
- Step 3: Validation in Isolated Mitochondria. Apply the constructed RNP-MITO-Porter to mitochondria isolated from target cells (e.g., HeLa cells or patient-derived fibroblasts). Incubate for one hour. Extract mtDNA and evaluate sequence-specific double-strand breaks using a comparative CT quantitative PCR method, comparing the amplification of the target region to a non-target mtDNA region [52].
- Step 4: In Vitro and In Vivo Application. Apply the RNP-MITO-Porter to target cells in culture. Confirm mitochondrial localization via confocal laser scanning microscopy using fluorescently labeled components. For in vivo application, further formulation may be required for systemic stability. Evaluate the reduction in mutant mtDNA heteroplasmy through droplet digital PCR or deep sequencing [52].
Navigating Safety and Genotoxicity in LNP-Mediated CRISPR Delivery
The therapeutic application of CRISPR-LNP complexes must contend with potential genotoxic risks, which extend beyond off-target editing at sites with sequence similarity to the gRNA.
- Structural Variations and Chromosomal Rearrangements: A pressing safety concern is the generation of large, on-target structural variations (SVs) following CRISPR-induced DSBs. These include kilobase- to megabase-scale deletions, chromosomal translocations, and rearrangements like chromothripsis [14]. Such SVs are particularly exacerbated by strategies that inhibit the non-homologous end joining (NHEJ) pathway, such as using DNA-PKcs inhibitors to enhance HDR. One study reported an "alarming thousand-fold increase" in the frequency of chromosomal translocations with such inhibitors [14] [21].
- Limitations of Standard Analysis: Common short-read amplicon sequencing can significantly underestimate these risks, as large deletions that remove primer-binding sites become "invisible." This can lead to an overestimation of HDR efficiency and a false sense of security regarding editing precision [14].
- Mitigation Strategies: A comprehensive safety assessment for clinical translation must include SV-detection methods like CAST-Seq or LAM-HTGTS [14]. Furthermore, the choice of CRISPR modality matters; while high-fidelity Cas9 variants and nickase-based systems reduce off-target effects, they can still introduce substantial on-target SVs. The field is moving towards a more nuanced risk-benefit analysis, acknowledging that for some diseases, even moderate editing levels may suffice without the need for HDR-enhancing strategies that carry greater genotoxic risk [14].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for LNP Development and CRISPR Delivery
| Reagent / Material | Function | Example Use Case |
|---|---|---|
| Ionizable Cationic Lipids | Core component for nucleic acid encapsulation and endosomal escape. | PL32 lipid for high-efficiency lung delivery [48]. |
| DSPC (Helper Lipid) | Provides structural integrity to the LNP bilayer. | Standard component in LNP formulations for stability [49]. |
| DMG-PEG2000 | Stabilizes particles and reduces nonspecific interactions. | Standard component to prevent aggregation and extend circulation time [49]. |
| STR-R8 (Stearylated-octaarginine) | Enhances cellular and mitochondrial uptake. | Key component in MITO-Porter for mitochondrial membrane fusion [52]. |
| Anti-PECAM-1 Antibody | Targeting ligand for lung endothelium. | Conjugated to LNP surface for redirected pulmonary delivery [50]. |
| Ursolic Acid | Natural product that activates V-ATPase. | Incorporated as a fifth component in NIF-LNPs to boost expression and reduce reactogenicity [48]. |
| Microfluidic Device | Enables reproducible, homogeneous LNP production. | iLiNP device for aseptic preparation of RNP-MITO-Porter [52]. |
Lipid nanoparticle technology has evolved from a simple encapsulation system to a sophisticated and programmable platform capable of directing CRISPR-Cas9 machinery to specific tissues and organelles following systemic administration. Breakthroughs in architecture, such as SNA designs, and in targeting, through Ab-LNP conjugates, are directly addressing the long-standing delivery challenges that have limited the clinical potential of gene editing. As these technologies mature, the focus will increasingly shift toward standardizing scalable production, comprehensively understanding long-term safety profiles, and expanding the repertoire of targetable tissues. The continued synergy between LNP innovation and CRISPR biology is poised to unlock a new frontier of precise, in vivo genetic medicines for a broad spectrum of diseases.
The CRISPR-Cas9 system has revolutionized biomedical research by providing an adaptable and precise method for generating targeted double-strand breaks (DSBs) in genomic DNA. Derived from a bacterial adaptive immune system, the platform consists of two fundamental components: a Cas9 nuclease and a guide RNA (gRNA) that directs the nuclease to a specific DNA sequence via Watson-Crick base pairing [53] [54]. Upon binding, Cas9 induces a DSB at the target site, activating the cellular DNA damage response machinery. The repair of these breaks primarily occurs through one of two major pathways: error-prone non-homologous end joining (NHEJ), which often results in small insertions or deletions (indels) that disrupt gene function, or homology-directed repair (HDR), which allows for precise genetic modifications using a donor DNA template [53] [54]. This fundamental mechanism—creating and then relying on cellular repair of DSBs—forms the cornerstone of all current CRISPR-based therapeutic applications.
As the field has matured, clinical pipelines have expanded beyond initial ex vivo applications to include innovative in vivo strategies, demonstrating promising results across a spectrum of diseases. This review provides a technical examination of three key therapeutic areas: hATTR amyloidosis, hereditary angioedema (HAE), and cancer, framing these advances within the core context of CRISPR-Cas9 mediated DSB formation and repair.
CRISPR-Cas9 Mechanism: From DSB Formation to Repair Pathways
Core Mechanism and DNA Repair Outcomes
The therapeutic efficacy of CRISPR-Cas9 is directly governed by the cellular response to the DSBs it generates. The following diagram illustrates the fundamental mechanism and the primary repair pathways that determine the genetic outcome.
The diagram above outlines the critical juncture after DSB formation. Most therapeutic applications currently leverage the NHEJ pathway to disrupt disease-causing genes. The HDR pathway, while offering precision, is inherently less efficient in most therapeutically relevant human cells [53]. This repair pathway dichotomy is central to understanding the design and outcome of the therapies discussed in this review.
Advanced DNA Repair Mechanisms and Experimental Manipulation
Beyond the classic NHEJ and HDR pathways, recent research has identified other repair mechanisms, such as CRISPR–homology-mediated end joining (HMEJ), which operates through a single-strand annealing process and shows high efficiency for gene targeting [54]. Furthermore, researchers often manipulate these repair pathways to achieve desired outcomes. For instance, inhibiting key NHEJ components like DNA-PKcs with small molecules (e.g., AZD7648) can shift the balance toward HDR [53]. However, such manipulations carry risks; DNA-PKcs inhibition has been shown to exacerbate genomic aberrations, including large kilobase- to megabase-scale deletions and increased frequencies of chromosomal translocations [53]. This underscores the complex trade-off between editing efficiency and genomic safety when interfering with the native DNA repair machinery.
Clinical Application I: hATTR Amyloidosis
Disease Mechanism and Therapeutic Strategy
Hereditary transthyretin (ATTR) amyloidosis is a monogenic disorder caused by mutations in the TTR gene, leading to the production of misfolded transthyretin protein that accumulates as amyloid fibrils in peripheral nerves, the cardiovascular system, and other organs [55]. This results in progressive neuropathy and/or cardiomyopathy [55]. The therapeutic strategy for hATTR amyloidosis involves using CRISPR-Cas9 to introduce a DSB in the TTR gene within hepatocytes, the primary site of TTR production. The subsequent repair via NHEJ results in disruptive indels that knockout the gene, thereby reducing the production of both mutant and wild-type TTR protein [55] [56].
Experimental Protocol and Clinical Workflow
The leading investigational therapy, NTLA-2001, utilizes an in vivo editing approach. The following workflow details the protocol from component preparation to patient administration and efficacy assessment.
Table 1: Key Research Reagents for hATTR Amyloidosis CRISPR Therapy
| Reagent / Component | Function in Experimental Protocol | Therapeutic Example |
|---|---|---|
| sgRNA targeting TTR | Guides Cas9 to a specific sequence within the human TTR gene to induce a DSB. | NTLA-2001 [55] |
| Cas9 mRNA | Encodes the Streptococcus pyogenes Cas9 nuclease protein; translated upon delivery into hepatocytes. | NTLA-2001 [55] |
| Liver-Tropic LNP | Biodegradable lipid nanoparticle that encapsulates CRISPR components and delivers them specifically to hepatocytes via APOE-LDL receptor mediated endocytosis. | NTLA-2001 [55] |
| Formulation Buffers | Maintain the stability and integrity of the LNP formulation during storage and administration. | Standard pharmaceutical excipients |
Quantitative Clinical Trial Data
Early-phase clinical trials have demonstrated substantial and durable knockdown of TTR protein levels with a single dose of NTLA-2001.
Table 2: Clinical Outcomes from hATTR Amyloidosis CRISPR Therapy
| Trial Phase | Dose | TTR Reduction (Mean) | Duration of Effect | Safety Profile |
|---|---|---|---|---|
| Phase 1 [55] | 0.1 mg/kg | 52% | At day 28 | Minimal, mild adverse events in 50% of participants. |
| Phase 1 [55] | 0.3 mg/kg | 87% | At day 28 | Transient increase in D-dimer in 83% of patients, resolved by day 7. |
| Phase 1/2 (Neuropathy) [8] | ~ 0.3 mg/kg (Projected) | ~90% | Sustained for 2+ years | Generally well-tolerated; mild or moderate infusion-related reactions common. |
| Phase 1/2 (Cardiomyopathy) [8] | ~ 0.3 mg/kg (Projected) | ~90% | Sustained for 2+ years | Generally well-tolerated; mild or moderate infusion-related reactions common. |
Clinical Application II: Hereditary Angioedema (HAE)
Disease Mechanism and Therapeutic Strategy
Hereditary angioedema (HAE) is a rare genetic disorder characterized by severe and unpredictable swelling attacks. It is driven by mutations in the SERPING1 gene, leading to overactivity of the plasma kallikrein-kinin system and excessive production of bradykinin, a potent vasodilator [57]. The investigational therapy NTLA-2002 employs a similar in vivo LNP-based strategy as NTLA-2001 but targets the KLKB1 gene, which encodes for prekallikrein. Knocking out KLKB1 via NHEJ-mediated repair reduces the production of plasma kallikrein, thereby preventing the pathological attacks [57].
Experimental Protocol and Clinical Workflow
The protocol for NTLA-2002 mirrors that of NTLA-2001, with modifications to the gRNA target and dosing specifics. The workflow involves LNP formulation with KLKB1-specific gRNA, intravenous infusion, hepatocyte delivery, on-target editing, and subsequent reduction in kallikrein protein and HAE attacks.
Quantitative Clinical Trial Data
Recent Phase 2 trial results for NTLA-2002 have shown remarkable efficacy in reducing attack rates.
Table 3: Clinical Outcomes from Hereditary Angioedema (HAE) CRISPR Therapy
| Trial Phase | Dose | Attack Rate Reduction (vs Placebo) | Patients Attack-Free (Wk 1-16) | Kallikrein Reduction |
|---|---|---|---|---|
| Phase 2 [57] | 25 mg | -75% | 4 of 10 (40%) | -55% at week 16 |
| Phase 2 [57] | 50 mg | -77% | 8 of 11 (73%) | -86% at week 16 |
| Phase 2 (Placebo) [57] | Placebo | - | 0 of 6 (0%) | No change |
The therapy was generally well-tolerated, with the most common adverse events being headache, fatigue, and nasopharyngitis [57]. These results demonstrate robust proof-of-concept for in vivo CRISPR-based intervention in HAE.
Clinical Application III: Cancer Immunotherapy
Therapeutic Strategy and Workflow
In oncology, CRISPR-Cas9 is primarily applied ex vivo to engineer a patient's own or donor-derived T cells for adoptive cell therapy. The dominant strategy involves creating allogeneic or autologous Chimeric Antigen Receptor (CAR) T cells. This process involves knocking out endogenous T-cell receptors (e.g., TRAC) and/or immune checkpoint genes (e.g., PD-1) to enhance potency and prevent graft-versus-host disease, while simultaneously knocking in a CAR transgene to direct T cells against tumor-specific antigens [58].
Experimental Protocol
The experimental protocol for generating CRISPR-edited CAR-T cells is a multi-step, ex vivo process.
Table 4: Key Research Reagents for Cancer CAR-T Cell CRISPR Engineering
| Reagent / Component | Function in Experimental Protocol | Therapeutic Example |
|---|---|---|
| CRISPR/Cas9 System | Creates DSBs at specific genomic loci (e.g., TRAC, PDCD1) to disrupt gene function or facilitate CAR knock-in. | CTX112 (Anti-CD19 CAR-T) [58] |
| CAR HDR Template | A donor DNA vector containing the CAR transgene, flanked by homology arms to guide its precise integration into a safe harbor or specific locus via HDR. | Various CAR-T therapies [58] |
| T Cell Media & Cytokines | Supports the activation (e.g., using anti-CD3/CD28 beads) and ex vivo expansion of T cells post-editing. | Standard cell culture reagents |
| Electroporation System | Enables efficient delivery of CRISPR ribonucleoproteins (RNPs) and HDR templates into primary T cells. | Clinical-grade electroporators |
Safety Considerations and Technical Challenges
A critical aspect of translating CRISPR-Cas9 therapies is understanding and mitigating the risks associated with unintended genomic alterations. Beyond the long-recognized concern of off-target (OT) mutagenesis at sites with sequence similarity to the target, recent studies reveal a more pressing challenge: large on-target structural variations (SVs) [53]. These include kilobase- to megabase-scale deletions, chromosomal translocations, and chromothripsis, which are particularly aggravated in cells treated with DNA-PKcs inhibitors used to enhance HDR [53]. Furthermore, traditional analytical methods like short-read amplicon sequencing can miss these large deletions if they span primer-binding sites, leading to an overestimation of successful HDR rates and an underestimation of indels and other aberrant outcomes [53]. The field is addressing these challenges through improved detection methods (e.g., CAST-Seq, LAM-HTGTS) and the development of more precise editing systems, such as prime editing and base editing, which can modify DNA without inducing DSBs, thereby minimizing the risk of SVs [54].
The clinical translation of CRISPR-Cas9 technology, grounded in the fundamental biology of DSB formation and repair, is demonstrating transformative potential across a growing spectrum of diseases. The success of in vivo therapies for hATTR amyloidosis and HAE, built upon targeted gene knockout via NHEJ and advanced LNP delivery, marks a pivotal advancement. Simultaneously, the sophisticated ex vivo engineering of CAR-T cells for oncology highlights the versatility of combining NHEJ-mediated knockout with HDR-mediated knock-in. As the clinical pipeline continues to expand, ongoing research into DSB repair mechanisms, coupled with rigorous safety profiling and the development of next-generation editors that avoid DSBs entirely, will be paramount. These efforts will ensure that CRISPR-based therapies can be applied with greater precision, efficacy, and safety, ultimately fulfilling their promise to treat and potentially cure a wide array of human diseases.
The CRISPR-Cas9 system has revolutionized genetic engineering by providing a precise and programmable method for creating double-strand breaks (DSBs) in DNA. This core mechanism—where the Cas9 nuclease is guided by RNA to specific genomic sequences—initiates cellular DNA repair processes that can be harnessed for therapeutic purposes [59]. Beyond well-established gene correction approaches, two advanced applications are emerging: the engineering of bacteriophages for antimicrobial therapy and the induction of large-scale genomic rearrangements for functional studies and potential treatments. Both strategies rely on fundamental principles of CRISPR-Cas9 but apply them to dramatically different biological contexts—from targeting bacterial pathogens to reorganizing complex eukaryotic genomes. This technical guide explores the mechanisms, methodologies, and applications of these novel strategies within the broader framework of CRISPR-mediated DSB formation and repair.
Phage Therapy Engineering via CRISPR-Cas Systems
Core Principles and Mechanisms
Bacteriophages (phages) are viruses that specifically infect and lyse bacterial cells, making them promising therapeutic agents against multidrug-resistant pathogens [60]. CRISPR-Cas systems enhance phage therapy by enabling precise genome engineering of phages to optimize their therapeutic properties. The type II CRISPR system with Cas9 is particularly valuable for this purpose, utilizing a guide RNA (gRNA) that contains a spacer sequence complementary to the target DNA and a scaffold sequence that enables Cas9 binding [60]. The Cas9-gRNA complex creates DSBs at specific locations in the phage genome, which are then repaired through cellular machinery to introduce targeted modifications.
Engineering phages addresses two major limitations of natural phage therapy: limited host range and the laborious process of phage discovery [60]. By modifying tail fibers and other receptor-binding proteins through CRISPR-mediated editing, phage host ranges can be broadened to target multiple bacterial species. Furthermore, CRISPR systems can introduce reporter genes (e.g., for fluorescent proteins) to facilitate the identification of successfully engineered phages [60].
Key Experimental Protocols
CRISPR-Cas9 Assisted Phage Engineering via Homologous Recombination:
- Design of Repair Template: Create a DNA insert containing the desired gene or modification flanked by homology arms (typically 500-1000 bp) identical to sequences upstream and downstream of the target site in the phage genome [60].
- Cloning and Transformation: Clone the insert into a replicative plasmid and transform it into a bacterial host strain susceptible to the target phage.
- Phage Infection and Recombination: Infect the transformed host with the wild-type phage. Homologous recombination occurs between the phage genome and the plasmid-based repair template.
- Selection and Screening: Identify recombinant phages by selecting for incorporated reporter genes (e.g., fluorescence) or through PCR-based verification of the modified genomic region [60].
Table 1: Key Reagents for CRISPR-Assisted Phage Engineering
| Reagent | Function | Specification Notes |
|---|---|---|
| Cas9 Nuclease | Creates DSBs at target sites | Can be delivered as protein or encoded on a plasmid [60]. |
| Guide RNA (gRNA) | Targets Cas9 to specific phage genomic loci | Requires complementary spacer to target sequence [60]. |
| Homology-Directed Repair (HDR) Template | Donor DNA for introducing modifications | Must contain desired change flanked by homology arms [60]. |
| Susceptible Bacterial Host | Provides cellular machinery for phage replication and recombination | Strain must be compatible with both phage infection and CRISPR plasmid maintenance [60]. |
| Selection Markers | Enriches for successfully engineered phages | Fluorescent proteins or antibiotic resistance genes [60]. |
Clinical Applications and Considerations
Phage therapy is particularly relevant for treating periprosthetic joint infections (PJIs), where biofilms formed by pathogens like Staphylococcus aureus and Pseudomonas aeruginosa confer significant antibiotic resistance [61]. Engineered phages can disrupt these biofilms through the production of depolymerizing enzymes. Clinical case studies have demonstrated the successful use of phage cocktails, administered via local injection or via antibiotic-impregnated bone cement, to treat PJIs after joint arthroplasty [61].
A prospective clinical study compared 23 PJI patients receiving adjunctive phage therapy with 22 historical controls treated only with antibiotics. The results indicated an eight times higher relapse rate in the control group after one year, suggesting phage therapy's potential to reduce infection recurrence [61]. Treatment was generally well-tolerated, with only mild, transient side effects reported.
Large-Scale Genomic Rearrangements via CRISPR-Cas9
Underlying Principles and Mechanisms
While CRISPR-Cas9 is often used for precise, small-scale edits, it can also induce large-scale genomic rearrangements—including deletions, duplications, inversions, and translocations—by generating multiple concurrent DSBs across the genome [53] [62]. The cellular repair of these breaks via error-prone non-homologous end joining (NHEJ) can result in the joining of non-adjacent DNA ends, leading to significant structural variations.
A method termed "Chromosome Rearrangement by CRISPR-Cas9 (CReaC)" has been developed to induce global chromosome rearrangement (GCR) [62]. This approach involves designing sgRNAs to target highly repetitive genomic elements, such as LINE-1 (L1) and Alu retrotransposons, which are dispersed throughout the human genome. Simultaneous cleavage at these numerous sites results in a large number of DSBs, triggering extensive genomic reshuffling [62].
Key Experimental Protocols
Inducing Global Chromosome Rearrangements with CReaC:
- sgRNA Design: Design sgRNAs targeting conserved regions of repetitive elements. For example, the sgRNA
TTCCAATCAATAGAAAAAGAtargets LINE-1 (L1), andTGTAATCCCAGCACTTTGGGtargets Alu elements [62]. - Vector Construction and Delivery: Clone sgRNA sequences into a CRISPR plasmid vector (e.g., pSB-CRISPR) and transfect into human cells (e.g., HEK293T) using lipid-based transfection reagents [62].
- Selection and Expansion: Select transfected cells with antibiotics (e.g., puromycin) for several weeks to establish polyclonal cell populations that have undergone rearrangement and survived.
- Karyotype and Genomic Analysis:
Safety Considerations: It is critical to assess the genotoxic risks of large-scale editing. Techniques like CAST-Seq and LAM-HTGTS can detect unforeseen structural variations and chromosomal translocations, which are critical for evaluating the safety of therapeutic editing approaches [53].
Table 2: Quantitative Analysis of CRISPR-Induced Structural Variations
| Genomic Alteration Type | Detection Method | Reported Frequency / Extent | Influencing Factors |
|---|---|---|---|
| Kilobase-scale deletions | Long-read WGS | Extensive in human stem cells [53] | Use of DNA-PKcs inhibitors (e.g., AZD7648) [53] |
| Megabase-scale deletions | Long-read WGS | Observed in multiple human cell types [53] | Inhibition of NHEJ pathway [53] |
| Chromosomal translocations | CAST-Seq, LAM-HTGTS | Frequency increased 1000-fold with NHEJ inhibition [53] | Simultaneous cutting at off-target sites [53] |
| Global CNV patterns | WGS, G-banding | Resemble patterns in tumor genomes [62] | Targeting of repetitive elements (LINE-1, Alu) [62] |
Research Applications and Biological Impact
The CReaC method enables the study of how large-scale genomic rearrangements influence cellular function and disease. In HEK293T cells, GCR induced by targeting L1 and Alu elements led to profound changes in the transcriptomic and epigenetic landscapes, altering pathways related to p53 signaling, DNA repair, cell cycle, and apoptosis [62]. These models provide valuable systems for understanding how chromosomal abnormalities contribute to diseases like cancer.
Integrated Workflows and Visualization
The experimental workflows for phage engineering and genomic rearrangement share a common foundation in CRISPR-Cas9 mechanics but diverge in their specific applications and outcomes. The following diagrams illustrate the core pathways and workflows for these two strategies.
CRISPR-Cas9 Mechanism in Non-Dividing Cells
Phage Engineering via CRISPR-HDR
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for CRISPR-Based Therapeutic Strategies
| Reagent Category | Specific Examples | Research Function |
|---|---|---|
| CRISPR Nucleases | Cas9, Cas12a, ARCUS nucleases [63] | Induces targeted DSBs for genome editing. Engineered variants offer improved specificity or novel cleavage patterns (e.g., 3' overhangs) [63]. |
| Delivery Systems | Virus-like particles (VLPs) [39], Lipid Nanoparticles (LNPs) [63], AAVs [59] | Enables efficient transport of CRISPR components into target cells (e.g., neurons, lymphocytes). |
| HDR Enhancers | Alt-R HDR Enhancer Protein [63], DNA-PKcs inhibitors [53] | Increases efficiency of precise homology-directed repair; requires careful safety profiling for genomic alterations [53]. |
| Phage Engineering Tools | Homologous recombination templates, CRISPR-Cas9 counter-selection systems [60] | Facilitates precise modification of bacteriophage genomes to alter host range or enhance therapeutic properties. |
| Analytical & Safety Tools | CAST-Seq [53], LAM-HTGTS [53], Long-read WGS [62] | Detects and characterizes structural variations, translocations, and other complex genomic outcomes of CRISPR editing. |
The applications of CRISPR-Cas9 in phage engineering and large-scale genomic rearrangements demonstrate the technology's expanding utility beyond single-gene editing. Both fields are progressing toward clinical translation, with phage therapy showing promise in combating multidrug-resistant infections [61], and chromosome rearrangement techniques providing powerful models for studying genomic instability [62].
Future development will focus on enhancing precision and safety. In phage therapy, this includes optimizing delivery systems and broadening host ranges without compromising specificity [60] [61]. In chromosomal engineering, critical challenges include mitigating on-target structural variations and understanding the long-term functional consequences of large-scale rearrangements [53] [62]. As CRISPR-based therapies advance, rigorous safety assessment using advanced genomic tools will be paramount for their successful translation into clinical applications [53].
Overcoming Technical Hurdles: Strategies for Enhancing Precision and Efficiency
The CRISPR-Cas9 system has revolutionized genome editing by enabling precise DNA double-strand breaks (DSBs) at targeted genomic loci. The system functions as an RNA-guided nuclease, where a Cas enzyme complexes with a guide RNA (gRNA) to identify and cleave DNA sequences complementary to the gRNA's spacer region, adjacent to a Protospacer Adjacent Motif (PAM) [2] [64]. Despite its transformative impact, a significant challenge impeding clinical translation is off-target activity (OTA)—unintended cleavage at sites with sequence similarity to the intended target [65] [64]. These off-target edits can confound experimental results and pose substantial safety risks in therapeutic contexts, including the potential for oncogenic transformations if edits occur in tumor suppressor genes or oncogenes [66] [67].
The tolerance of the CRISPR-Cas9 system for mismatches between the gRNA and DNA target is a primary source of OTA. Wild-type Streptococcus pyogenes Cas9 (SpCas9) can tolerate between three and five base pair mismatches, particularly in the PAM-distal region of the target site [2] [66]. Furthermore, the system's specificity is influenced by cellular factors, including chromatin accessibility and the DNA repair pathway engaged following the DSB [2] [64]. The error-prone non-homologous end joining (NHEJ) pathway often repairs these breaks, leading to insertions or deletions (indels) that can disrupt gene function [2] [68]. This review details strategies to mitigate off-target effects, focusing on high-fidelity Cas variants and optimized gRNA design, framed within the mechanistic context of DSB formation and repair.
The Core Mechanism of CRISPR-Cas9 and Origins of Off-Target Effects
A thorough understanding of the CRISPR-Cas9 mechanism is essential for developing effective mitigation strategies. The process begins with the formation of a ribonucleoprotein complex between the Cas9 nuclease and a single-guide RNA (sgRNA). This complex surveys the genome, searching for a short PAM sequence (5'-NGG-3' for SpCas9) [2]. PAM recognition triggers local DNA melting, allowing the ~20-nucleotide spacer sequence of the gRNA to form a heteroduplex with the target DNA strand through Watson-Crick base pairing [2] [64].
If complementarity is sufficient, particularly in the 8-12 nucleotide "seed region" adjacent to the PAM, a stable R-loop structure forms. This induces a conformational change in Cas9, activating its two nuclease domains: the HNH domain, which cleaves the target DNA strand, and the RuvC-like domain, which cleaves the non-target strand [2]. This action typically results in a blunt-ended or slightly staggered DSB [2].
The cellular machinery then repairs the DSB primarily through one of two major pathways. The first is the accurate, template-dependent Homology-Directed Repair (HDR), which can be co-opted for precise gene correction but is relatively rare in somatic cells [2] [68]. The second is the more common but error-prone Non-Homologous End Joining (NHEJ), which directly ligates the broken ends, often introducing small insertions or deletions (indels) [2]. Additional pathways like Microhomology-Mediated End Joining (MMEJ) can also contribute to mutagenic outcomes [2] [64]. Off-target effects occur when this sophisticated machinery is deceived by sequences similar to the intended target, leading to DSBs at unintended genomic locations. The following diagram illustrates the key steps in this process and the subsequent repair choices.
Diagram: CRISPR-Cas9 Mechanism and Off-Target Origin. The diagram illustrates the sequence of events from Cas9-gRNA complex formation to DNA cleavage and repair, highlighting how mismatch tolerance can lead to off-target effects via the error-prone NHEJ pathway.
Strategies for Mitigating Off-Target Effects
High-Fidelity Cas9 Variants
A primary strategy to reduce OTA involves engineering or discovering Cas nucleases with enhanced specificity. While wild-type SpCas9 is effective, its promiscuity has driven the development of high-fidelity variants with reduced tolerance for gRNA-DNA mismatches. These enzymes are typically engineered through rational design or directed evolution to introduce point mutations that destabilize the Cas9-gRNA-DNA complex in the presence of mismatches [66] [67].
The table below summarizes key high-fidelity SpCas9 variants, their engineering strategies, and their primary characteristics.
Table 1: High-Fidelity Cas9 Variants and Their Properties
| Variant Name | Engineering Strategy | Key Characteristics | Considerations |
|---|---|---|---|
| SpCas9-HF1 [67] | Rational design of four mutations to reduce non-specific interactions with the DNA phosphate backbone. | Dramatically reduced off-target activity; maintains robust on-target efficiency for many targets. | On-target efficiency can be variable and guide-dependent. |
| eSpCas9(1.1) [67] | Engineered to increase the energy threshold for DNA cleavage, requiring more perfect guide-target complementarity. | Effective suppression of off-target cleavage across multiple targets. | May exhibit reduced on-target activity in some contexts. |
| HypaCas9 [66] [67] | Comprehensive structure-guided engineering for hyper-accurate DNA recognition. | Superior specificity profile; enhanced fidelity without compromising on-target activity in many cases. | Requires empirical validation for each gRNA. |
| evoCas9 [67] | Directed evolution in yeast to select for variants that minimize cleavage at mismatched targets. | Functionally evolved for high specificity; demonstrates strong on-target to off-target performance. | Like others, performance is not universal across all genomic loci. |
It is critical to note that while these high-fidelity variants reduce off-target cleavage, they do not necessarily reduce off-target binding [66] [67]. This distinction is vital for applications using catalytically dead Cas9 (dCas9) for transcriptional regulation or epigenome editing, where DNA binding alone can have functional consequences. Furthermore, the trade-off for enhanced specificity can sometimes be reduced on-target editing efficiency, necessitating careful empirical optimization for each application [66].
Guide RNA (gRNA) Design and Optimization
The selection and design of the gRNA are equally critical for minimizing OTA. An optimal gRNA maximizes on-target cleavage while minimizing potential interactions with off-target sites [64] [66].
Key gRNA Design Parameters:
- Sequence Uniqueness: The primary design goal is to select a spacer sequence with minimal homology to other genomic sites. Software tools like CRISPOR, Cas-OFFinder, and CCTop are used to scan the genome for potential off-target sites and rank gRNA candidates based on their predicted specificity scores [66] [67].
- GC Content: Guides with moderate to high GC content (40-60%) tend to form more stable DNA:RNA duplexes, which can improve on-target efficiency. However, very high GC content may increase the risk of off-target binding [66].
- Chemical Modifications: Synthetic gRNAs can be chemically modified to enhance stability and specificity. Common modifications include incorporating 2'-O-methyl analogs (2'-O-Me) and 3' phosphorothioate bonds (PS). These alterations can protect the gRNA from nuclease degradation and, crucially, alter the thermodynamics of binding to favor on-target over off-target interactions [2] [66].
- Truncated gRNAs: Shortening the gRNA spacer sequence from 20 to 17-18 nucleotides can reduce its stability and thereby decrease its tolerance for mismatches, lowering off-target activity, though this may also reduce on-target efficiency [66].
- Double Nickase Strategy: Using a pair of gRNAs with Cas9 nickase (nCas9), which creates single-strand breaks instead of DSBs, can dramatically improve specificity. A DSB is only formed when two nicks occur in close proximity on opposite strands. The probability of this happening at an off-target site is vastly lower than at the intended on-target site [67].
Table 2: Summary of gRNA Optimization Strategies
| Strategy | Method | Impact on Specificity | Practical Considerations |
|---|---|---|---|
| In Silico Design | Use of algorithms (e.g., CRISPOR) to select guides with unique sequences and high specificity scores. | Foundation of specificity; prevents avoidable off-targets. | First and most critical step in experimental design. |
| Chemical Modification | Adding 2'-O-Me and PS bonds to synthetic gRNAs. | Reduces off-target editing and improves nuclease resistance. | Cost factor for synthetic guides; requires titration for efficacy. |
| Length Modification | Using truncated gRNAs (tru-gRNAs) of 17-18 nt. | Increases specificity by reducing binding energy. | Can significantly reduce on-target efficiency. |
| Dual gRNA Nicking | Employing two adjacent gRNAs with a Cas9 nickase (D10A mutant). | Very high specificity, as DSB requires two independent binding events. | Requires two efficient gRNAs; potential for larger deletions. |
Experimental Protocols for Off-Target Assessment
Rigorous assessment of off-target activity is mandatory for therapeutic development and high-stakes research. The following protocols outline standardized methods for this critical analysis.
In Silico Prediction and Candidate Site Sequencing
This is the most common initial approach for off-target assessment.
- Primary Tools: CRISPOR, CCTop, Cas-OFFinder.
- Procedure:
- Input your candidate gRNA sequence and the relevant reference genome into the prediction tool.
- The algorithm generates a list of potential off-target sites, typically ranked by a score that considers the number and position of mismatches and bulges.
- Select the top ~10-20 predicted off-target sites for experimental validation.
- Design PCR primers to amplify these genomic regions from edited and control samples.
- Perform next-generation sequencing (NGS) of the amplified products.
- Analyze the sequencing data using tools like the Inference of CRISPR Edits (ICE) or other variant callers to quantify the frequency of indels at each candidate site [66] [67].
- Advantages: Low cost and straightforward.
- Limitations: Relies on the accuracy of prediction algorithms and may miss off-target sites with low sequence homology.
Genome-Wide Off-Target Detection Methods
For a more comprehensive analysis, especially in clinical applications, genome-wide methods are required.
- GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing):
- During CRISPR delivery, a short, double-stranded oligonucleotide tag is transfected into cells.
- This tag is incorporated into CRISPR-induced DSBs via the NHEJ repair pathway.
- Genomic DNA is sheared and sequenced using NGS.
- Reads containing the tag sequence are mapped back to the genome to identify all sites of DSB formation, both on-target and off-target [66] [67].
- Circle-seq:
- Genomic DNA is extracted and circularized in vitro.
- The circularized DNA is treated with Cas9-gRNA complexes under cleavage-favorable conditions.
- DNA that is cleaved (at on- or off-target sites) becomes linearized.
- The linearized fragments are purified and sequenced, providing a sensitive, cell-free profile of potential cleavage sites [66].
- Whole Genome Sequencing (WGS):
- Perform deep (>50x coverage) WGS on clonal cell lines derived from edited cells or on bulk edited populations.
- Compare the genomes to unedited control cells using sophisticated bioinformatics pipelines to identify de novo mutations, including single-nucleotide variants (SNVs), indels, and larger structural variations (SVs) [66] [69].
- WGS is considered the gold standard for comprehensive off-target assessment as it does not rely on a priori assumptions about potential off-target loci [69].
The following diagram illustrates a typical experimental workflow that integrates these assessment methods.
Diagram: Off-Target Assessment Workflow. The flowchart outlines a tiered strategy for off-target assessment, from initial in silico prediction to comprehensive genome-wide sequencing, depending on the application's requirements.
Successfully executing a specific and safe CRISPR-Cas9 experiment requires a suite of specialized reagents and tools. The table below details essential components for mitigating off-target effects.
Table 3: Research Reagent Solutions for Specific CRISPR Editing
| Reagent / Tool | Function | Example & Notes |
|---|---|---|
| High-Fidelity Cas9 Expression Vector | Delivers the gene for a high-specificity nuclease (e.g., SpCas9-HF1, eSpCas9). | Plasmids or mRNA for in vitro transcription; available from Addgene and commercial vendors. |
| Chemically Modified Synthetic gRNA | Enhances gRNA stability and specificity. | Synthego and other suppliers offer gRNAs with 2'-O-Me and PS modifications. |
| Cas9 Nickase (D10A) | Enables the dual gRNA nicking strategy for high-specificity DSB formation. | Available as plasmid, mRNA, or protein from standard commercial sources. |
| Off-Target Prediction Software | Identifies potential off-target sites during gRNA design. | CRISPOR (web tool), Cas-OFFinder (web tool). Critical for initial guide selection. |
| Off-Target Detection Kits | Wet-bench kits for genome-wide off-target identification. | Commercial GUIDE-seq or CIRCLE-seq kits (e.g., from Integrated DNA Technologies). |
| Analysis Software | Quantifies editing efficiency and identifies indels from sequencing data. | ICE (Inference of CRISPR Edits) for Sanger data; various NGS pipelines for deeper analysis. |
Mitigating the off-target effects of CRISPR-Cas9 is not a single-step task but a comprehensive strategy integral to its therapeutic and research applications. As detailed in this guide, this involves a multi-pronged approach: selecting high-fidelity Cas variants like eSpCas9(1.1) or HypaCas9, rigorously optimizing gRNA design with the aid of computational tools and chemical modifications, and employing robust experimental protocols such as GUIDE-seq or WGS for thorough off-target profiling [65] [64] [66]. The choice of delivery method (e.g., RNP complexes) to limit the duration of nuclease activity also plays a crucial role [66]. By systematically applying these principles, researchers can significantly enhance the specificity of CRISPR-Cas9-mediated DSB formation, thereby unlocking its full potential for precise genetic manipulation and safe human therapeutics.
The CRISPR-Cas9 system has revolutionized biomedical research and therapeutic development by enabling precise manipulation of the genome. The core mechanism involves the creation of a site-specific double-strand break (DSB) in DNA, which is subsequently repaired by the cell's endogenous repair pathways [70]. The formation and repair of this DSB is the foundational event upon which all CRISPR-based editing depends. The most common repair pathways are error-prone non-homologous end joining (NHEJ), which often results in insertions or deletions (indels) that disrupt the gene, and the more precise homology-directed repair (HDR), which can facilitate specific gene corrections [71] [72]. The initial step in this process is the efficient and safe delivery of the CRISPR-Cas9 machinery—typically comprising the Cas nuclease and a guide RNA (gRNA)—to the nucleus of target cells [73].
The transformative potential of genomic medicines can only be fully realized if the CRISPR system can be delivered to specific organs and cell types directly in vivo [71]. The choice of delivery vector is thus paramount, influencing everything from editing efficiency and specificity to immunogenicity and clinical feasibility. This whitepaper provides a technical analysis of the two predominant delivery strategies—viral vectors and lipid nanoparticles (LNPs)—focusing on their respective challenges and advances in achieving tissue-specific targeting within the context of CRISPR-Cas9 DSB research and therapy development.
Delivery Platforms: A Comparative Analysis
The delivery of CRISPR-Cas9 components can be achieved using viral vectors, synthetic non-viral systems like LNPs, or physical methods. The selection of a delivery platform involves trade-offs between payload capacity, efficiency, specificity, and safety [74].
Viral Vector Systems
Viral vectors leverage the natural ability of viruses to infect cells and are among the most efficient delivery vehicles used in gene therapy and editing.
- Adeno-Associated Viruses (AAVs): AAVs are a leading platform for in vivo CRISPR delivery due to their low immunogenicity, high transduction efficiency in diverse tissues, and ability to sustain long-term transgene expression. Different AAV serotypes exhibit natural tropisms for specific tissues, such as AAV8 and AAV9 for the liver, which can be harnessed for targeting [71].
- Lentiviral Vectors (LVs): LVs are capable of integrating into the host genome, enabling stable, long-term expression of the CRISPR machinery. This makes them particularly suitable for ex vivo applications, such as engineering chimeric antigen receptor (CAR) T-cells [71] [72].
- Adenoviral Vectors (AVs): AVs can accommodate large DNA payloads and achieve high transduction efficiency, but their use is limited by pre-existing immunity in human populations and strong inflammatory responses [71].
A primary constraint of viral vectors, especially AAVs, is their limited packaging capacity (~4.7-5 kb), which is insufficient for the canonical Streptococcus pyogenes Cas9 (SpCas9). This has driven the development and use of smaller Cas9 orthologues, such as Staphylococcus aureus Cas9 (SaCas9) and Campylobacter jejuni Cas9 (cjCas9) [71]. Furthermore, immunogenicity remains a concern, as pre-existing or treatment-induced immune responses against the viral capsid can neutralize the vector and prevent re-dosing, a significant hurdle for chronic diseases [71] [74].
Non-Viral Systems: Lipid Nanoparticles (LNPs)
LNPs are synthetic, self-assembling particles composed of ionizable lipids, phospholipids, cholesterol, and lipid-anchored polyethylene glycol (PEG). They have emerged as a powerful non-viral delivery platform, famously validated by their success in delivering mRNA vaccines. For CRISPR-Cas9, LNPs can efficiently encapsulate and deliver Cas9 mRNA and sgRNA, or pre-assembled Cas9-gRNA ribonucleoproteins (RNPs) [71] [70].
A key advantage of LNP delivery, particularly with RNP formats, is the transient presence of the nuclease inside the cell. This minimizes off-target editing and reduces the risk of immunogenic responses against the bacterial Cas9 protein. LNPs also offer a superior safety profile by avoiding the risk of genomic integration associated with some viral vectors. However, a major challenge has been the natural tropism of first-generation LNPs for the liver and spleen following systemic administration, limiting their application for extrahepatic diseases [71] [74]. Current research is focused on re-engineering LNP lipid compositions and surface functionalization with targeting ligands (e.g., antibodies, peptides) to redirect them to specific tissues [74].
Table 1: Quantitative Comparison of CRISPR-Cas9 Delivery Systems
| Feature | Adeno-Associated Virus (AAV) | Lentivirus (LV) | Lipid Nanoparticles (LNPs) |
|---|---|---|---|
| Payload Capacity | Limited (~4.7-5 kb) [71] | Large (~8 kb) [71] | High (suitable for mRNA/RNPs) [70] |
| Typical Payload | DNA encoding SaCas9/cjCas9 + sgRNA [71] | DNA encoding SpCas9 + sgRNA [71] | Cas9/sgRNA mRNA or RNP complexes [71] [70] |
| Immunogenicity | Moderate to High (capsid/transgene immunity) [71] [74] | Moderate [71] | Low (especially with RNP delivery) [70] |
| Integration Risk | Low (predominantly episomal) | High (random integration) | None |
| Manufacturing | Complex, high cost | Complex, moderate cost | Scalable, more cost-effective |
| Tropism/ Targeting | Serotype-dependent natural tropism; can be engineered [71] [74] | Broad tropism; pseudotyping possible | Innate liver/spleen tropism; targeting requires engineering [74] |
| Editing Duration | Long-term (stable expression) | Long-term (integration) | Short-term (transient activity) |
Experimental Protocols for In Vivo Delivery and Analysis
To guide researchers in evaluating delivery systems, the following are detailed protocols for systemic administration of AAV and LNP carriers, and for analyzing editing outcomes in complex in vivo models.
Protocol 1: Systemic Delivery of CRISPR-Cas9 via AAV Vectors for Liver Targeting
This protocol outlines the steps to achieve in vivo gene editing in the mouse liver using AAV8, a serotype with high hepatocyte tropism [71].
Vector Design and Packaging:
- Select a small Cas9 orthologue (e.g., SaCas9) compatible with AAV packaging constraints.
- Clone the expression cassette for SaCas9 and the target-specific sgRNA into an AAV plasmid backbone under the control of appropriate promoters (e.g., a liver-specific promoter for enhanced safety and efficacy).
- Package the recombinant genome into AAV8 capsids using a standard triple-transfection method in HEK293 cells, followed by purification via iodixanol gradient ultracentrifugation or affinity chromatography.
- Determine the viral genome titer (vg/mL) using quantitative PCR.
In Vivo Administration:
- Animals: Use adult C57BL/6 mice (or a disease-relevant model, such as the mdx mouse for Duchenne muscular dystrophy [71]).
- Injection: Adminify the AAV8 vector via tail vein injection at a typical dose of 1x10^11 to 1x10^12 vector genomes (vg) per mouse in a 100-200 µL sterile saline solution [71].
- Controls: Include a control group injected with a non-targeting sgRNA or PBS.
Tissue Harvest and Analysis:
- Timeline: Euthanize animals at the experimental endpoint (e.g., 2-4 weeks post-injection) and harvest the liver.
- Genomic DNA Extraction: Homogenize a section of the liver and extract genomic DNA using a commercial kit.
- Editing Efficiency Assessment:
- Amplify the target genomic region by PCR.
- Quantify indel formation using the T7 Endonuclease I (T7E1) mismatch cleavage assay or by next-generation sequencing (NGS) for a more accurate and quantitative readout.
Figure 1: AAV-mediated CRISPR delivery workflow for liver editing.
Protocol 2: Internally Controlled Screening with CRISPR-StAR in Tumors
Conventional pooled CRISPR screens in complex in vivo models like tumors are confounded by bottlenecks in cell engraftment and heterogeneous clonal outgrowth, which introduce massive noise [75]. The CRISPR-StAR (Stochastic Activation by Recombination) method overcomes this by generating internal controls within each single-cell-derived clone.
Library and Cell Line Preparation:
- Clone a sgRNA library into the CRISPR-StAR vector backbone. This vector uses intercalated loxP and lox5171 sites to create two mutually exclusive recombination outcomes upon Cre exposure: one producing an active sgRNA and the other an inactive one [75].
- Generate a stable Cas9-expressing tumor cell line (e.g., mouse melanoma cells) and transduce it with the CRISPR-StAR library at a high coverage (>500 cells/sgRNA). Use a cell line that also expresses a tamoxifen-inducible Cre recombinase (Cre-ERT2).
In Vivo Tumor Formation and Induction:
- Transplant the transduced cell pool into immunodeficient or immunocompetent mice to form tumors.
- Once tumors are established, administer tamoxifen to activate Cre-ERT2. This stochastically converts the sgRNA library within the engrafted, expanded clones into a mosaic of active and inactive states, creating the internal control for each clone [75].
Analysis and Hit Calling:
- After a period of tumor progression (e.g., 2-3 weeks), harvest the tumors and recover the sgRNA sequences.
- Use next-generation sequencing to quantify the abundance of each active sgRNA and its corresponding inactive control within every unique molecular identifier (UMI)-tagged clone.
- Calculate the fold-change depletion or enrichment of each active sgRNA relative to its own internal control within the same clone. This intra-clonal normalization corrects for variability in clonal expansion, dramatically improving signal-to-noise ratio and hit-calling accuracy compared to conventional analysis [75].
Figure 2: CRISPR-StAR workflow for high-resolution genetic screening in vivo.
The Scientist's Toolkit: Essential Research Reagents
Successful execution of CRISPR delivery experiments requires a suite of specialized reagents and tools. The following table details key solutions for research in this field.
Table 2: Key Research Reagent Solutions for CRISPR-Cas9 Delivery Studies
| Research Reagent | Function and Application |
|---|---|
| AAV Serotypes (e.g., AAV8, AAV9, AAV6) | Leverage innate tissue tropisms: AAV8/9 for liver and heart; AAV9 for crossing blood-brain barrier; AAV6 for muscle [71]. |
| Ionizable LNPs | Synthetic carriers that encapsulate and protect CRISPR payloads (mRNA, RNPs); enable efficient cellular uptake and endosomal escape [70]. |
| Cre-ERT2 Inducible System | Allows precise, tamoxifen-dependent control of genetic recombination; essential for methods like CRISPR-StAR to temporally control sgRNA activation [75]. |
| Unique Molecular Identifiers (UMIs) | Short random nucleotide sequences used to barcode individual cells or clones; enables clonal tracking and lineage tracing in complex pools and in vivo models [75]. |
| T7 Endonuclease I (T7E1) Assay | A rapid, cost-effective method for initial quantification of indel efficiency at the target genomic locus by detecting and cleaving DNA heteroduplex mismatches. |
| Next-Generation Sequencing (NGS) | The gold standard for comprehensive analysis of editing outcomes, providing precise quantification of indels, HDR efficiency, and unbiased detection of off-target effects. |
| Anti-CRISPR Proteins (Acrs) | Natural inhibitors of Cas nucleases; used as chemical control tools to rapidly turn off Cas9 activity after editing, minimizing off-target effects [70]. |
The field of CRISPR delivery is rapidly evolving to overcome the dual challenges of efficiency and specificity. Future directions include the integration of artificial intelligence (AI) to design novel tissue-specific LNPs and to predict the optimal Cas9 variant and gRNA combination for a given therapeutic target [74]. Furthermore, the development of "smart" bioresponsive materials that release their CRISPR payload only upon encountering specific cellular signals (e.g., tumor-specific enzymes) holds promise for enhancing specificity [70]. Beyond inducing DSBs, newer CRISPR modalities like base editing and prime editing offer more precise genetic alterations without requiring DSBs, but their larger size presents an even greater delivery challenge, further incentivizing the development of advanced viral and non-viral vectors [74].
In conclusion, both viral vectors and LNPs present distinct advantages and formidable challenges for CRISPR-Cas9 delivery. Viral vectors offer high efficiency and durability, but are constrained by packaging limits and immunogenicity. LNPs provide a transient, potentially safer profile with greater packaging flexibility, but require significant engineering to escape hepatic sequestration and target extrahepatic tissues. The choice of delivery system is ultimately dictated by the specific application, target tissue, and desired duration of editing. Overcoming the delivery bottleneck through continued innovation in vector design and targeting strategies remains the critical path forward for translating CRISPR-Cas9 genome editing into widespread clinical reality.
The CRISPR-Cas9 system has revolutionized biological research and therapeutic development by enabling precise genome editing. This process initiates with the formation of a site-specific double-strand break (DSB) in the DNA, catalyzed by the Cas9 nuclease guided by a single guide RNA (sgRNA) [76]. The cellular response to this DSB is a critical determinant of the editing outcome, primarily governed by two competing repair pathways: error-prone non-homologous end joining (NHEJ) and high-fidelity homology-directed repair (HDR) [76] [28].
Achieving precise genomic modifications via HDR represents a central challenge in genome engineering, particularly because NHEJ is the predominant and more efficient repair pathway in most cellular contexts, especially post-mitotic cells [76]. The balance between these pathways is influenced by multiple factors including cell type, cell cycle stage, nuclease platform, and target locus [77]. This technical guide examines the molecular mechanisms underlying DSB repair pathway choice and provides evidence-based strategies for manipulating this balance to enhance HDR efficiency, with particular relevance for research and therapeutic applications.
DNA Repair Pathway Mechanisms
Non-Homologous End Joining (NHEJ)
NHEJ is an error-prone repair mechanism that functions throughout the cell cycle and represents the dominant DSB repair pathway in mammalian cells [76] [23]. This pathway initiates with the recognition of DSB ends by the Ku heterodimer complex (Ku70/Ku80), which subsequently recruits and activates DNA-dependent protein kinase catalytic subunit (DNA-PKcs) [76]. The Artemis nuclease is then activated to process DNA ends, followed by ligation by the XRCC4-DNA ligase IV complex [76]. NHEJ often results in small insertions or deletions (indels) that disrupt gene function, making it particularly useful for gene knockout studies [28].
Homology-Directed Repair (HDR)
HDR is a precise repair mechanism that utilizes a homologous DNA template to faithfully restore sequence information at the break site [76]. Unlike NHEJ, HDR is restricted primarily to the S and G2 phases of the cell cycle when a sister chromatid is available as a natural template [23]. In experimental settings, researchers can provide an exogenous donor template containing homologous arms flanking the desired modification. The process involves resection of DNA ends to create 3' single-stranded overhangs, invasion of the homologous template, and synthesis-dependent repair [76]. While HDR enables precise gene correction and knock-in approaches, its efficiency is typically substantially lower than NHEJ across most biological contexts [77].
Quantitative Analysis of HDR and NHEJ Efficiencies
Systematic quantification of repair pathway outcomes reveals substantial variability depending on experimental conditions. Using a novel droplet digital PCR (ddPCR) assay capable of simultaneously detecting HDR and NHEJ events, researchers have demonstrated that the HDR/NHEJ ratio is highly dependent on gene locus, nuclease platform, and cell type [77]. Contrary to the prevailing assumption that NHEJ generally occurs more frequently than HDR, certain conditions can actually yield more HDR than NHEJ [77].
Table 1: HDR and NHEJ Efficiencies Across Nuclease Platforms in HEK293T Cells
| Nuclease Platform | Target Locus | HDR Efficiency (%) | NHEJ Efficiency (%) | HDR/NHEJ Ratio |
|---|---|---|---|---|
| Wildtype Cas9 | RBM20 | 16.5 ± 1.8 | 25.3 ± 2.1 | 0.65 |
| Cas9 D10A Nickase | RBM20 | 13.7 ± 1.5 | 8.6 ± 0.9 | 1.59 |
| FokI-dCas9 | RBM20 | 12.9 ± 1.2 | 6.3 ± 0.7 | 2.05 |
| TALEN | RBM20 | 10.8 ± 1.1 | 5.2 ± 0.6 | 2.08 |
| Wildtype Cas9 | GRN | 22.4 ± 2.3 | 29.8 ± 3.0 | 0.75 |
| Cas9 D10A Nickase | GRN | 19.6 ± 2.0 | 12.1 ± 1.3 | 1.62 |
Table 2: Cell Type-Dependent Variation in HDR and NHEJ Efficiencies
| Cell Type | Nuclease Platform | Target Locus | HDR Efficiency (%) | NHEJ Efficiency (%) | HDR/NHEJ Ratio |
|---|---|---|---|---|---|
| HEK293T | Wildtype Cas9 | RBM20 | 16.5 ± 1.8 | 25.3 ± 2.1 | 0.65 |
| HeLa | Wildtype Cas9 | RBM20 | 8.3 ± 0.9 | 31.2 ± 3.2 | 0.27 |
| Human iPSCs | Wildtype Cas9 | RBM20 | 5.2 ± 0.6 | 18.7 ± 1.9 | 0.28 |
| HEK293T | Cas9 D10A Nickase | GRN | 19.6 ± 2.0 | 12.1 ± 1.3 | 1.62 |
| HeLa | Cas9 D10A Nickase | GRN | 9.8 ± 1.0 | 15.3 ± 1.6 | 0.64 |
Strategic Modulation of Repair Pathways
Inhibition of the NHEJ Pathway
Strategic inhibition of key NHEJ components can effectively shift the repair balance toward HDR. Small molecule inhibitors targeting specific NHEJ factors have demonstrated particular utility:
- DNA-PKcs Inhibitors: Compounds such as NU7026 and KU0060648 suppress NHEJ by blocking the activity of DNA-dependent protein kinase, a critical component of the NHEJ machinery [76].
- Ku Complex Interference: Molecular approaches that disrupt the Ku70/Ku80 heterodimer, the initial DSB sensor in NHEJ, can reduce error-prone repair [76].
Recent investigations into PARP1 modulation have revealed its significant influence on DSB repair pathway choice. PARP1 downregulation increases both NHEJ and microhomology-mediated end joining (MMEJ) without altering HDR efficiency, while PARP1 overexpression reduces NHEJ and HDR but leaves MMEJ unaffected [21]. This positions PARP1 as a key regulator of DSB repair balance and a potential target for improving editing precision.
Activation of the HDR Pathway
Enhancing the intrinsic HDR capacity of cells represents a complementary approach to improving precise genome editing outcomes:
- Small Molecule Enhancers: Compounds such as RS-1 (a RAD51 stimulator) and L755507 can increase HDR efficiency by promoting the activity of key homologous recombination factors [76].
- Cell Cycle Synchronization: Since HDR is primarily active in S/G2 phases, chemical synchronization using agents such as nocodazole (G2/M arrest) or mimosine (G1/S arrest) can enrich for HDR-competent cell populations [76].
- Temperature Modulation: Mild hypothermia (32°C) following editing has been shown to enhance HDR efficiency in some cell types, potentially by extending the timeframe for homologous recombination [76].
Nuclease Engineering and Selection
The choice of nuclease platform significantly influences repair pathway outcomes. Engineered Cas9 variants can reduce indel formation and favor precise editing:
- Cas9 Nickases: Cas9 D10A (RuvC domain mutation) creates single-strand nicks rather than DSBs. Using paired nickases targeting opposite strands can generate staggered DSBs with 5' overhangs that may be more amenable to HDR [76] [77].
- FokI-dCas9 Systems: Catalytically dead Cas9 fused to FokI nuclease domains requires dimerization for activity, enhancing specificity and potentially favoring HDR in certain contexts [77].
- High-Fidelity Cas9 Variants: Engineered Cas9 proteins with reduced off-target activity, such as eSpCas9 and SpCas9-HF1, may indirectly influence repair outcomes by minimizing unintended DSBs [78].
Experimental Protocols for Pathway Analysis
Droplet Digital PCR (ddPCR) for Simultaneous HDR and NHEJ Quantification
The ddPCR assay enables highly sensitive and simultaneous quantification of HDR and NHEJ events at endogenous loci, capable of detecting one HDR or NHEJ event per 1,000 genome copies [77].
Protocol:
- Design Principles: Amplicons should flank the nuclease cut site (positioned 3 bp upstream of PAM for CRISPR systems), with 75-125 bp on either side. For HDR detection, design one primer outside the donor molecule sequence to ensure quantification of integrated edits [77].
- Probe Design: Utilize dual-labeled fluorescent probes (FAM/HEX) with distinct emission spectra. A reference probe should be positioned distant from the cut site. In some cases, a dark, non-extendible oligonucleotide (3' phosphorylation) may be designed to block cross-reactivity of the HDR probe with wildtype sequences [77].
- Reaction Setup: Partition 20 μL reactions into 20,000 nanodroplets using a droplet generator. Thermal cycling conditions: 95°C for 10 min (enzyme activation), followed by 40 cycles of 94°C for 30 s (denaturation) and specific annealing temperature (determined empirically) for 60 s (extension), then 98°C for 10 min (enzyme deactivation) [77].
- Data Analysis: Quantify positive droplets using a droplet reader and analyze with companion software. Calculate editing efficiencies as the ratio of positive droplets for each event (HDR or NHEJ) to reference positive droplets [77].
Luminescent and Fluorescent Reporter Assays for Pathway Choice
Reporter systems enable rapid screening of factors influencing repair pathway balance, particularly useful for assessing chemical modulators or protein factors.
PARP1 Modulation Protocol [21]:
- Reporter Construction: Develop luminescent and fluorescent reporter constructs sensitive to specific repair pathways (NHEJ, MMEJ, HDR).
- Cell Transfection: Co-transfect reporter constructs with PARP1 modulation vectors (overexpression or knockdown) and CRISPR-Cas9 components.
- Pathway Assessment: Quantify repair outcomes by measuring luminescent/fluorescent signals 72 hours post-transfection.
- Validation: Confirm results by targeted sequencing of endogenous loci to correlate reporter data with actual genomic editing outcomes.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Manipulating DNA Repair Pathways
| Reagent Category | Specific Examples | Function/Mechanism | Application Context |
|---|---|---|---|
| NHEJ Inhibitors | NU7026, KU0060648 | DNA-PKcs inhibition | Enhances HDR efficiency by suppressing competing NHEJ pathway |
| HDR Enhancers | RS-1, L755507 | RAD51 stimulation | Promotes homologous recombination and strand invasion |
| Cell Cycle Synchronizers | Nocodazole, Aphidicolin | G2/M or G1/S arrest | Enriches HDR-competent cell populations |
| Nuclease Platforms | Cas9 D10A, FokI-dCas9 | Engineered specificity | Reduces indel formation; favors precise editing |
| Donor Templates | Single-stranded oligodeoxynucleotides (SSODNs) | Homology-directed repair template | Template for precise gene correction or insertion |
| Detection Tools | ddPCR assays, NGS | Quantitative outcome analysis | Simultaneous measurement of HDR and NHEJ events |
The strategic balancing of NHEJ versus HDR efficiency represents a cornerstone of precision genome engineering. The competing DNA repair pathways exhibit complex dependencies on experimental parameters including nuclease platform, target locus, cell type, and cell cycle status. By leveraging specific inhibitors, enhancers, and engineered nucleases, researchers can significantly shift the balance toward desired repair outcomes.
The ongoing development of more sophisticated quantification methods, particularly ddPCR and advanced reporter systems, continues to refine our understanding of these processes. As CRISPR-based therapies advance toward clinical application, with recent successes in rare genetic diseases like sickle cell anemia and transthyretin amyloidosis [8], precise control over DNA repair outcomes becomes increasingly critical. Future directions will likely include more temporal and spatial control over repair processes, as well as continued engineering of novel CRISPR systems that inherently favor precise editing.
CRISPR-Cas9-mediated homology-directed repair (HDR) represents a versatile platform for creating precise site-specific DNA insertions, deletions, and substitutions, holding tremendous promise for gene therapy, disease modeling, and functional genomics [76]. This precise editing mechanism relies on exogenous donor templates carrying desired sequences to guide repair at DNA break sites. However, a fundamental biological constraint severely limits its application: the inherently low efficiency of HDR in non-dividing or postmitotic cells [76] [79]. Unlike error-prone non-homologous end joining (NHEJ) that functions throughout the cell cycle, HDR is largely restricted to the S and G2 phases where sister chromatids are available as natural repair templates. This cell cycle dependency creates a significant barrier for therapeutic applications targeting quiescent or terminally differentiated cells, such as neurons, cardiomyocytes, and hematopoietic stem cells [79]. Overcoming this limitation requires a multifaceted understanding of the competing DNA repair pathways and innovative strategies to tilt the balance toward precise genome editing.
Biological Mechanisms: DNA Repair Pathway Competition
The persistence of HDR constraints in non-dividing cells stems from fundamental biological mechanisms of DNA repair pathway competition. When CRISPR-Cas9 induces double-strand breaks (DSBs), multiple repair pathways activate, with NHEJ dominating in postmitotic cells due to its template-independent nature and constant activity throughout the cell cycle [76].
Non-Homologous End Joining (NHEJ) Pathway
The NHEJ pathway represents the predominant DSB repair mechanism in mammalian cells, characterized by rapid activation without requirement for homologous templates. The process initiates with Ku protein complex recognition of broken DNA ends, where Ku70 and Ku80 subunits form a ring structure encircling the DNA [76]. This complex then recruits downstream repair proteins through three primary sub-pathways:
- Blunt-end ligation-dependent Ku-XRCC4-DNA ligase IV sub-pathway: Directly ligates broken ends after Cas9-mediated blunt-end cleavage [76]
- Nuclease-dependent sub-pathway: Involves DNA-PKcs recruitment and Artemis endonuclease activation for end processing before ligation [76]
- Polymerase-dependent sub-pathway: Engages Pol μ and Pol λ for fill-in synthesis of mismatched ends [76]
The constitutive activity of these NHEJ mechanisms throughout the cell cycle explains its dominance in non-dividing cells, where HDR machinery remains largely inaccessible.
Homology-Directed Repair (HDR) Pathway
HDR operates through fundamentally different mechanisms requiring homologous donor templates. In cycling cells, HDR progresses through resection of DNA ends to create 3' single-stranded overhangs, strand invasion with homologous templates, DNA synthesis using donor sequences, and resolution of recombination intermediates [76]. This process depends on Rad51 and other recombination proteins that show cell cycle-regulated expression and activity, with peak function during S and G2 phases when sister chromatids are available. The near-absence of these coordinated activities in non-dividing cells creates the fundamental barrier to HDR efficiency that necessitates strategic interventions.
Diagram: The competitive balance between NHEJ and HDR pathways favors NHEJ in non-dividing cells, creating the fundamental challenge for precise genome editing.
Quantitative Analysis of HDR Limitations
Comprehensive analysis of CRISPR-Cas9 editing outcomes reveals substantial quantitative challenges for HDR in biologically relevant systems. Recent studies utilizing advanced detection methods have quantified both the intended HDR events and unintended consequences that disproportionately affect non-dividing cells.
Efficiency Metrics Across Cell Types
Table 1: HDR Efficiency and Large Deletion Events in Primary Cells and Cell Lines
| Cell Type | Target Gene | HDR Efficiency Range | Large Deletion Frequency | Experimental System |
|---|---|---|---|---|
| Hematopoietic Stem/Progenitor Cells (HSPCs) | HBB | 11.7-35.4% | 11.7-35.4% | HiFi SpCas9 RNP [80] |
| Hematopoietic Stem/Progenitor Cells (HSPCs) | HBG | Not specified | 14.3% | HiFi SpCas9 RNP [80] |
| Hematopoietic Stem/Progenitor Cells (HSPCs) | BCL11A | Not specified | 13.2% | HiFi SpCas9 RNP [80] |
| Primary T Cells | PD-1 | Not specified | 15.2% | HiFi SpCas9 RNP [80] |
| S-HUDEP2 Cell Line | HBB | Variable by clone | High (clonal genotyping) | Single-cell derived clones [80] |
The data demonstrate that large deletion events represent a significant competing outcome that further reduces functional HDR efficiency. These substantial deletions (up to several thousand base pairs) occur with high frequencies at Cas9 on-target cut sites and persist through cell differentiation, potentially altering biological functions and reducing available therapeutic alleles [80].
Advanced Detection Methodologies
Accurate quantification of HDR constraints requires sophisticated methodologies beyond standard short-range sequencing:
- Long-amplicon sequencing (LongAmp-seq): Illumina NGS of fragmented long-range PCR products detecting both small INDELs and large deletions in one assay [80]
- SMRT-seq with unique molecular identifiers (UMI): Pacific Biosciences circular consensus sequencing with dual UMIs to mitigate PCR chimeras and length-dependent biases [80]
- Droplet digital PCR (ddPCR) allelic drop-off assay: Absolute quantification of deletion events without amplification bias [80]
- BreakTag profiling: Genome-wide mapping of Cas9-induced DSBs with end structure resolution at nucleotide resolution [81]
These methodologies reveal that conventional short-range NGS significantly underestimates the complexity of editing outcomes, particularly the substantial large deletion events that compete with successful HDR in therapeutically relevant primary cells.
Strategic Approaches to Enhance HDR Efficiency
Pathway Modulation Strategies
Multiple strategic approaches have been developed to overcome HDR constraints by modulating the competitive balance between DNA repair pathways:
NHEJ Inhibition
- Chemical inhibition of key NHEJ proteins (DNA-PKcs, Ku70/80)
- RNA interference against DNA ligase IV
- Dominant-negative mutants of NHEJ complex components [76]
HDR Pathway Activation
- Small molecule enhancers of key HDR factors (Rad51, BRCA1/2)
- Cell cycle synchronization to enrich for S/G2 populations
- Expression of Cas9 fusion proteins with HDR-promoting factors [76]
Donor Template Engineering
- Single-stranded DNA donors with optimized homology arm lengths
- Virus-derived templates (AAV) with high nuclear delivery efficiency
- Chemical modification of donor ends to resist nuclease degradation [76] [82]
Cas9 Reagent Delivery
- Ribonucleoprotein (RNP) complexes for rapid engagement and clearance
- Engineered Cas9 variants with biased cleavage profiles
- Modulation of Cas9 expression levels to limit persistent DSB formation [76] [81]
Experimental Workflow for HDR Optimization
Diagram: Comprehensive experimental workflow for assessing HDR efficiency and quantifying unintended editing outcomes in non-dividing cell systems.
Research Reagent Solutions
Table 2: Essential Research Reagents for HDR Optimization Studies
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| CRISPR Nucleases | HiFi SpCas9 [80], Cas9 D10A nickase [76], eSpCas9(1.1) | Engineered variants with reduced off-target effects and biased cleavage profiles |
| Donor Templates | ssODN (≤150 nt) [82], AAV donor vectors, dsDNA plasmid donors | Homology-containing repair templates with optimized arm length and chemical modifications |
| Pathway Modulators | DNA-PKcs inhibitors (KU-0060648), Rad51 agonists (RS-1), NHEJ pathway siRNAs | Chemical and genetic tools to shift repair balance toward HDR pathway |
| Detection Assays | LongAmp-seq reagents [80], BreakTag library prep [81], ddPCR assay kits | Specialized reagents for comprehensive editing outcome analysis beyond standard INDEL detection |
| Delivery Systems | Cas9 RNP complexes [80], Electroporation enhancers, Viral delivery vectors | Efficient delivery modalities for maximal editing with minimal persistent nuclease activity |
Addressing HDR constraints in non-dividing cells remains a critical challenge for therapeutic genome editing applications. The competition from efficient NHEJ pathways combined with the cell cycle regulation of HDR machinery creates a biological barrier that requires sophisticated multi-pronged approaches. Future research directions emphasize the development of high-precision Cas9 variants with biased cleavage profiles, HDR-independent precise editing tools, and improved delivery systems for transient nuclease activity [79] [81]. Additionally, comprehensive analysis of editing outcomes using long-read sequencing and DSB profiling technologies will provide crucial insights into the full spectrum of genetic modifications, enabling more accurate risk-benefit assessments for clinical applications. As these technologies mature, combined approaches targeting both the enhancement of HDR and suppression of competing repair pathways offer promising avenues for overcoming the fundamental limitation of HDR efficiency in therapeutically relevant non-dividing cell populations.
The CRISPR-Cas9 system has revolutionized genetic engineering by providing an efficient mechanism for targeted DNA cleavage. This system operates by creating double-strand breaks (DSBs) at specific genomic locations, relying on cellular repair processes to achieve desired genetic modifications. However, a growing body of evidence reveals that the therapeutic application of DSB-dependent editing is challenged by significant safety concerns, including the generation of complex, unintended genomic alterations [14].
Beyond well-documented off-target effects at sites with sequence similarity to the target site, DSB induction can lead to large structural variations (SVs), including chromosomal translocations, megabase-scale deletions, and chromothripsis [14]. These genotoxic events raise substantial safety concerns for clinical translation, particularly when edits occur in tumor suppressor genes or oncogenes. Furthermore, DSB repair pathways are cell cycle-dependent, with the preferred pathway for precise correction (homology-directed repair, HDR) largely restricted to the S and G2 phases, making precise editing inefficient in non-dividing cells [83].
These limitations have motivated the development of advanced engineering solutions that enable precise genetic modifications without inducing DSBs. Base editing and prime editing represent two groundbreaking technologies that fulfill this requirement, offering researchers and therapeutic developers powerful tools for precision genome engineering with enhanced safety profiles.
Base Editing: Programmable Chemical Conversion of Nucleotides
Molecular Architecture and Mechanism
Base editing enables the direct, irreversible chemical conversion of one DNA base pair to another without DSB formation. The technology utilizes a catalytically impaired Cas protein (either dead Cas9/dCas9 or nickase Cas9/nCas9) fused to a nucleobase deaminase enzyme [84]. The system operates through a sophisticated multi-step mechanism:
- Targeted Binding: The Cas protein, guided by a sgRNA, binds to the target DNA sequence without cleaving both strands [85].
- Local Strand Separation: The R-loop formation exposes a stretch of single-stranded DNA (ssDNA) to the deaminase enzyme [84].
- Nucleobase Deamination: The deaminase enzyme chemically modifies specific nucleobases within an "editing window" (typically 4-5 nucleotides in the spacer region) [86].
- Cellular Processing: The cell's endogenous DNA repair machinery recognizes and processes the intermediate, leading to permanent base conversion.
Two primary classes of base editors have been developed: Cytosine Base Editors (CBEs) convert C•G base pairs to T•A, while Adenine Base Editors (ABEs) convert A•T base pairs to G•C [84] [85].
Table 1: Major Base Editor Classes and Their Core Components
| Editor Class | Deaminase Enzyme | Cas Component | Key Accessory Proteins | Primary Conversion |
|---|---|---|---|---|
| Cytosine Base Editor (CBE) | Cytidine deaminase (e.g., rAPOBEC1, eA3A) | nCas9 (D10A) | Uracil glycosylase inhibitor (UGI) | C•G → T•A |
| Adenine Base Editor (ABE) | Evolved tRNA adenosine deaminase (eTadA) | nCas9 (D10A) | None required | A•T → G•C |
For CBEs, the deaminase converts cytosine to uracil within the ssDNA bubble, creating a U•G mismatch. The fused UGI inhibits uracil DNA glycosylase (UNG), preventing excision of the uracil and increasing editing efficiency [84]. The nickase activity of nCas9 on the non-edited strand induces cellular mismatch repair (MMR) to preferentially replace the G with an A, completing the C•G to T•A conversion [87].
ABEs operate through a similar mechanism but utilize an evolved TadA deaminase to convert adenine to inosine, which is treated as guanine by cellular polymerases. The resulting I•T mismatch is then resolved to G•C through MMR [84].
Experimental Workflow for Base Editing
A standard protocol for conducting base editing experiments in mammalian cells involves the following key steps:
- Target Selection and gRNA Design: Identify a target site with the desired base within the editing window (typically positions 4-8 within the protospacer, counting the PAM as positions 21-23) and ensure the presence of a compatible PAM sequence (NGG for SpCas9) [84].
- Editor Delivery:
- Plasmid Transfection: Co-transfect cells with plasmids encoding the base editor and sgRNA.
- Ribonucleoprotein (RNP) Delivery: Pre-complex the purified base editor protein with sgRNA and deliver via electroporation for reduced off-target effects [88].
- Viral Delivery: For in vivo applications, package split-intein base editor systems into dual AAV vectors due to size constraints [89].
- Analysis of Editing Outcomes:
- Harvest genomic DNA 48-72 hours post-editing.
- Amplify the target region by PCR and sequence using next-generation sequencing (NGS).
- Quantify the frequency of precise base conversion, indels, and bystander edits (unintended edits at nearby bases within the editing window).
Diagram 1: CBE Mechanism – From binding to permanent base conversion.
Research Reagent Solutions for Base Editing
Table 2: Essential Research Reagents for Base Editing Applications
| Reagent / Tool | Function | Example Variants / Formulations |
|---|---|---|
| Base Editor Proteins | Catalyzes targeted base conversion | BE4max, AncBE4max, ABEmax, ABE8e [84] |
| Guide RNA | Targets editor to specific genomic locus | sgRNA with optimized spacer sequence |
| Delivery Vehicles | Introduces editing components into cells | AAV vectors (split-intein), LNPs, electroporation [89] |
| Reporter Systems | Measures editing efficiency and outcomes | Fluorescent reporters (BFP-to-GFP, non-fluorescent GFP turn-on) [87] |
| Inhibitors/Modulators | Manipulates DNA repair to influence outcomes | UNG inhibitors, MMR modulators [87] |
Prime Editing: A Search-and-Replace Platform for Precision Editing
Molecular Architecture and Mechanism
Prime editing represents a monumental leap in precision genome editing, functioning as a "search-and-replace" system that directly writes new genetic information into a specified DNA site. This versatile technology can mediate targeted insertions, deletions, and all 12 possible base-to-base conversions without requiring DSBs or donor DNA templates [90].
A prime editor consists of three core components:
- A Cas9 nickase (H840A) that cleaves only the DNA strand containing the protospacer adjacent motif (PAM) sequence.
- An engineered reverse transcriptase (RT) that synthesizes DNA using an RNA template.
- A prime editing guide RNA (pegRNA) that both specifies the target site and encodes the desired edit through its 3' extension, which contains a primer binding site (PBS) and an RT template (RTT) [90] [86].
The multi-step prime editing mechanism proceeds as follows:
- Targeted Strand Nicking: The prime editor complex binds to the target DNA, and the Cas9 nickase cleaves the PAM-containing strand.
- Reverse Transcription: The 3' end of the nicked DNA hybridizes to the PBS of the pegRNA and primes reverse transcription using the RTT, generating a flap containing the edited sequence.
- Flap Resolution and Ligation: Cellular enzymes resolve the branched intermediate by excising the original 5' flap and ligating the edited 3' flap into the genome.
- Strand Correction (Optional): In the PE3 system, an additional sgRNA directs nicking of the non-edited strand to bias cellular repair toward permanent incorporation of the edit [90] [86].
Diagram 2: PEn Editing Pathways – Dual repair mechanisms for insertion.
Experimental Workflow for Prime Editing
A comprehensive protocol for prime editing implementation includes:
- pegRNA Design:
- Design the spacer sequence (typically 20 nt) to target the desired locus with an appropriate PAM.
- Define the RT template (RTT, ~10-16 nt) to encode the desired edit, ensuring it does not hybridize with the spacer.
- Include a PBS (~8-15 nt) that is complementary to the DNA flanking the nick site but does not contain sequences that could self-hybridize with the RTT.
- Stability Enhancement:
- Incorporate structured RNA motifs (e.g., evopreQ1, mpknot) at the 3' end of the pegRNA to create engineered pegRNAs (epegRNAs) that resist degradation and improve editing efficiency [86].
- Editor Delivery:
- Transfert cells with plasmids encoding the prime editor (PE2) and pegRNA.
- For enhanced efficiency, co-deliver an additional nicking sgRNA (PE3 system) to direct nicking of the non-edited strand.
- Utilize viral vectors (e.g., dual AAV systems for the split prime editor, sPE) for in vivo applications [86].
- Outcome Analysis:
- Extract genomic DNA 3-7 days post-editing.
- Amplify the target region and analyze by NGS, specifically employing methods capable of detecting large deletions that might be missed by short-read amplicon sequencing [14].
- Quantify the percentage of precise intended edits, indels, and other byproducts.
Advanced Prime Editing Systems
Recent engineering efforts have significantly enhanced prime editing capabilities:
- PE2: Features an engineered M-MLV reverse transcriptase with five mutations that enhance thermostability, processivity, and affinity for RNA-DNA hybrids, improving editing efficiency over the original PE1 [86].
- PE3: Incorporates an additional sgRNA to nick the non-edited strand, encouraging the cellular repair machinery to use the edited strand as a template, thereby increasing editing efficiency [90] [86].
- PEn (Prime Editor nuclease): Utilizes wild-type SpCas9 nuclease (instead of a nickase) to create a DSB, harnessing homology-dependent repair or non-homologous end joining (NHEJ) to install edits. This approach can rescue inefficiently edited sites with standard PE2/PE3 systems [83].
Table 3: Quantitative Comparison of Major Genome Editing Platforms
| Editing Technology | Editing Capabilities | DSB Formation? | Typical Efficiency Range | Key Limitations |
|---|---|---|---|---|
| CRISPR-Cas9 Nuclease | Gene disruption (indels) | Yes | High (often >80% indels) | Unpredictable repair outcomes, large deletions, translocations [14] |
| Cytosine Base Editor (CBE) | C•G → T•A transitions | No | Moderate to High (often 10-50%) | Restricted to specific transitions, bystander edits, off-target deamination [84] |
| Adenine Base Editor (ABE) | A•T → G•C transitions | No | Moderate to High (often 10-50%) | Restricted to specific transitions, larger size challenges delivery [84] |
| Prime Editor (PE2/PE3) | All point mutations, small insertions/deletions | No | Variable (1-50%, highly site-dependent) | Efficiency can be low, complex pegRNA design [90] [86] |
Base editing and prime editing technologies represent a paradigm shift in precision genome engineering, offering powerful alternatives to conventional DSB-dependent CRISPR-Cas9 systems. The strategic selection between these platforms depends on the specific research or therapeutic objective:
- Base editors are ideal for efficient and precise correction of single-nucleotide polymorphisms involving transition mutations (C•G→T•A or A•T→G•C), with particular utility in diseases where even partial correction provides therapeutic benefit [89].
- Prime editors provide unparalleled versatility, capable of installing all possible base substitutions, small insertions, and deletions, making them suitable for addressing a broader spectrum of pathogenic mutations, including transversions and small indels [90] [86].
As these technologies continue to evolve through protein engineering, optimized delivery strategies, and enhanced understanding of cellular repair mechanisms, they are poised to overcome current limitations in efficiency and targeting scope. Their demonstrated success in preclinical models and ongoing progression toward clinical trials solidifies base editing and prime editing as foundational tools for the next generation of precision genetic medicines, effectively addressing the critical safety concerns associated with DSB formation while significantly expanding the scope of therapeutically actionable mutations.
CRISPR-Cas9 in Context: Validation Methods and Platform Comparisons
The CRISPR-Cas9 system has revolutionized biomedical research and therapeutic development by enabling precise genome editing. This technology utilizes a Cas9 nuclease and a single-guide RNA (sgRNA) to create targeted DNA double-strand breaks (DSBs), which are then repaired by cellular mechanisms such as non-homologous end joining (NHEJ) or homology-directed repair (HDR) [13] [28]. However, a significant challenge compromising the precision of this system is the occurrence of off-target effects—unintended edits at genomic sites with sequence similarity to the target site, often due to tolerable mismatches between the sgRNA and DNA [91] [92]. These off-target mutations can potentially lead to adverse consequences, including the activation of oncogenes or disruption of vital genes, posing a substantial risk for clinical applications [91].
Therefore, rigorous analytical validation of editing outcomes is not merely a best practice but a fundamental requirement. This involves a suite of methods designed to predict, detect, and quantify off-target activity. As emphasized in the FDA's guidance for CRISPR-based therapies, a combination of multiple methods, including genome-wide analysis, is recommended to ensure comprehensive off-target profiling [93]. This whitepaper provides an in-depth technical guide to the primary methods for off-target detection, with a particular focus on the role of whole-genome sequencing (WGS) as a critical, unbiased tool for analytical validation.
A Landscape of Off-Target Detection Methodologies
Off-target detection strategies can be broadly categorized into computational predictions, in vitro biochemical assays, and in vivo cellular assays. The following table summarizes the core approaches, while subsequent sections detail key protocols.
Table 1: Overview of Off-Target Detection Approaches
| Approach | Examples | Principle | Sensitivity | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| In Silico (Biased) | Cas-OFFinder, CCTop, CCLMoff [13] [94] | Algorithms predict sites based on sgRNA sequence homology and PAM rules. | N/A | Fast, inexpensive; ideal for initial sgRNA design and screening. | Relies on reference genomes; may miss sgRNA-independent or variant-associated sites [93]. |
| Biochemical (Unbiased) | Digenome-seq, CIRCLE-seq, SITE-seq [91] [93] | Cas9 cleaves purified genomic DNA in vitro; breaks are sequenced. | Very High (can detect ultra-rare sites) | Ultra-sensitive; works with any DNA sample; no cellular context needed. | May overestimate off-targets due to lack of chromatin and cellular repair machinery [93]. |
| Cellular (Unbiased) | GUIDE-seq, DISCOVER-seq [13] [93] | DSBs in living cells are captured via tagged oligos or repair protein binding. | High | Captures edits in a physiological context with native chromatin and repair. | Requires efficient delivery into cells; may miss very low-frequency events [93]. |
| In Situ (Unbiased) | BLESS, BLISS [91] [13] | DSBs are labeled and captured in fixed cells, preserving nuclear architecture. | Moderate | Preserves genomic architecture; captures breaks in situ. | Technically complex; lower throughput; single time-point snapshot [93]. |
| Whole-Genome Sequencing | N/A [95] [96] | High-throughput sequencing of the entire genome of edited and control cells. | Varies with coverage | Truly genome-wide and hypothesis-free; detects all variant types (SNPs, indels, structural variations). | Expensive; requires deep sequencing for sensitivity; complex data analysis; requires a reference genome [96]. |
In Silico Prediction Tools
Computational tools form the first line of defense against off-target effects. Early tools like Cas-OFFinder and CCTop perform genome-wide scanning to identify sites with sequence homology to the sgRNA, allowing for a user-defined number of mismatches and bulges [13]. More advanced, learning-based models like CCLMoff have emerged, which use deep learning frameworks and pretrained RNA language models to capture mutual sequence information between sgRNAs and target sites. These models are trained on comprehensive datasets from multiple experimental methods, enabling more accurate prediction and better generalization across different genomic contexts [94].
Key Experimental Workflows
GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) This cellular method identifies off-target DSBs directly in living cells.
- Transfection: Cells are co-transfected with the Cas9/sgRNA RNP complex and a double-stranded oligodeoxynucleotide (dsODN) tag [13] [93].
- Tag Integration: The dsODN tag is preferentially integrated into CRISPR-induced DSBs by the cell's endogenous repair machinery.
- Genomic DNA Extraction & Library Prep: Genomic DNA is sheared and prepared for next-generation sequencing (NGS). Adapters are ligated, and PCR amplification is performed using primers specific to the integrated dsODN tag.
- Sequencing & Analysis: High-throughput sequencing identifies genomic locations flanking the integrated tag, revealing the landscape of both on-target and off-target DSBs [93].
CIRCLE-seq (Circularization for In vitro Reporting of Cleavage Effects by Sequencing) This highly sensitive biochemical method detects potential off-target sites in vitro.
- DNA Extraction & Circularization: Genomic DNA is purified and enzymatically circularized [94] [93].
- In Vitro Cleavage: Circularized DNA is treated with Cas9/sgRNA RNP complexes to induce DSBs at sites recognized by the complex.
- Enrichment & Linearization: An exonuclease digests linear, non-cleaved DNA, enriching the pool for cleaved fragments. The remaining circular DNA is then linearized.
- Sequencing & Analysis: The linearized DNA fragments, which represent Cas9 cleavage sites, are prepared into a sequencing library and analyzed via NGS [93].
DISCOVER-seq (Discovery of In Situ Cas Off-Targets by Verification and Sequencing) This cellular method leverages the natural DNA repair response to identify off-targets.
- Editing & Cell Fixation: Living cells are edited with CRISPR-Cas9. At a specific time point post-editing, cells are fixed.
- Immunoprecipitation: Chromatin is sheared and an antibody against the DNA repair protein MRE11 is used to pull down DNA fragments associated with active repair sites [13] [93].
- Sequencing & Analysis: The enriched DNA is sequenced, mapping the genomic loci where Cas9-induced breaks are being actively repaired by the cell [93].
The Confirmatory Role of Whole-Genome Sequencing
While the above methods are powerful for discovery, whole-genome sequencing (WGS) serves as a critical, unbiased tool for definitive analytical validation. WGS involves sequencing the entire genome of edited and control cells at high depth, allowing for the identification of all variant types—SNPs, indels, and larger structural variations—without any prior assumptions about their location [95] [96].
Protocol for Off-Target Assessment via WGS:
- Sample Preparation: Genomic DNA is extracted from multiple CRISPR-edited cell lines or organisms and from wild-type controls [96].
- Library Construction & Sequencing: High-quality DNA is used to construct sequencing libraries. These libraries are then sequenced on a high-throughput platform to achieve sufficient coverage (e.g., 30x-50x or higher) for confident variant calling [96].
- Bioinformatic Analysis:
- Alignment: Clean sequencing reads are aligned to a reference genome.
- Variant Calling: Computational tools are used to call SNPs and indels by comparing the edited samples to the wild-type controls.
- Off-Target Filtering: The massive list of identified variants is filtered to isolate true off-targets. This involves:
- Focusing on variants located at computationally predicted off-target sites.
- Excluding common genetic variants and background mutations present in the cell line or strain.
- Validating candidate off-target indels via an orthogonal method, such as Sanger sequencing [96].
Evidence from Research: A WGS study in grapevine underscored the high specificity of CRISPR/Cas9. The researchers sequenced seven edited and three wild-type plants, identifying hundreds of thousands of natural variants. However, after rigorous filtering of potential off-target sites, only a single, validated off-target indel mutation was discovered, demonstrating that careful sgRNA design can result in minimal off-target activity [96].
The following diagram illustrates the logical relationship and application of these methods within a comprehensive analytical validation workflow.
Diagram 1: Off-target analysis workflow. This shows a staged strategy from prediction to definitive validation.
Successful off-target analysis relies on a suite of specialized reagents and tools. The table below details key components.
Table 2: Essential Research Reagents and Tools for Off-Target Analysis
| Item | Function | Application Examples |
|---|---|---|
| Cas9 Nuclease (WT & High-Fidelity) | Creates DSBs at target DNA sites. High-fidelity variants (e.g., eSpCas9, SpCas9-HF1) are engineered to reduce off-target cleavage [91]. | All experimental detection methods. |
| sgRNA Synthesis Reagents | Produces the guide RNA that directs Cas9 to a specific genomic locus. | All experimental detection methods. |
| Lipid Nanoparticles (LNPs) | A delivery vehicle for in vivo CRISPR-Cas9 components, with a natural tropism for the liver [8]. | Enables in vivo off-target studies in animal models. |
| Double-Stranded ODN Tag | A short, double-stranded DNA oligo that integrates into DSBs for genome-wide mapping [93]. | GUIDE-seq. |
| Anti-MRE11 Antibody | Binds to the MRE11 DNA repair protein to immunoprecipitate DNA fragments at active repair sites [93]. | DISCOVER-seq. |
| Biotinylated Adaptors / Cas9 | Labels DSB ends for streptavidin-based enrichment and sequencing. | BLESS, SITE-seq [91] [93]. |
| Next-Generation Sequencer | Provides the high-throughput sequencing capability required for genome-wide analysis. | WGS, GUIDE-seq, CIRCLE-seq, etc. [95] |
The safe and effective translation of CRISPR-Cas9 technology into therapies hinges on rigorous analytical validation of its precision. A tiered strategy that integrates computational prediction, highly sensitive biochemical discovery, biologically relevant cellular validation, and confirmatory whole-genome sequencing provides the most robust framework for off-target assessment. As the field advances, the development of more sensitive cellular assays, standardized protocols by bodies like NIST, and AI-driven predictive models will further enhance our ability to ensure the safety of this transformative technology [94] [93].
The efficacy of CRISPR-Cas9 genome editing is fundamentally governed by the cellular response to the programmable double-strand break (DSB). While the mechanism of DSB formation by the Cas9 nuclease is well-characterized, the subsequent repair by cellular machinery is highly variable, making the prediction of editing outcomes a significant challenge. Recent research has revealed that editing efficiency and fidelity are not solely properties of the CRISPR system itself, but are profoundly influenced by the cellular context, including cell type, genetic background, and the specific sequence being targeted [97]. This technical guide explores the current frameworks for quantifying CRISPR-Cas9 editing efficacy, establishes critical benchmarks across diverse experimental conditions, and provides detailed methodologies for researchers to accurately measure and interpret these outcomes within the broader thesis of DSB formation and repair.
The initial assumption that Cas9-induced breaks are repaired randomly has been superseded by evidence showing strong sequence context-dependence [81]. Furthermore, the type of Cas9 incision—whether it results in a blunt end or a staggered end with overhangs—has been linked to distinct repair outcomes, with staggered breaks associated with more predictable, single-nucleotide insertions [81]. This establishes a direct mechanistic link between the biophysics of the DSB formation and the final editing product, a core concept for this discussion.
Core Concepts: Defining Editing Efficiency and Outcome Diversity
Key Metrics and Terminology
- Editing Efficiency: Typically reported as the percentage of alleles within a cell population that contain any modification (indel) at the target site. This is a primary metric for knockout generation.
- Homology-Directed Repair (HDR) Efficiency: The percentage of alleles that have successfully incorporated a desired precise edit using a donor DNA template.
- Indel Spectrum: The distribution of different insertion and deletion mutations resulting from NHEJ repair. A predictable spectrum is desirable for reproducible knockout models.
- Kinetic Rate Constants (k): Quantitative descriptors of the DSB repair process, including the cutting rate (k~c~), perfect repair rate (k~p~), and mutagenic repair rate (k~m~) [97].
- Structural Variations (SVs): Large, unintended genomic alterations such as megabase-scale deletions, translocations, or chromosomal rearrangements that are critical safety benchmarks [14] [98].
The DSB Repair Kinetic Model
Quantitative insights into DSB repair at single loci have been achieved using kinetic models that treat the process as a three-state system (Figure 1) [97]. In this model, the DNA is either intact, broken, or has been repaired with an indel. The rate constants governing the transitions between these states determine the final editing outcome.
Figure 1. Kinetic Model of Cas9-Induced DSB Repair. The model defines three DNA states and the rate constants (k~c~, k~p~, k~m~) for transitions between them. The "Indel DNA" state is irreversible as the modified sequence is typically no longer recognized by the sgRNA.
Studies applying this model have revealed that repair of Cas9-induced DSBs is often slow and error-prone, with half-lives of the broken state lasting up to ~10 hours, and a significant portion of breaks being resolved via mutagenic pathways [97].
Quantitative Benchmarks Across Cell Types and Loci
The efficiency and fidelity of CRISPR-Cas9 editing are not uniform. The following tables summarize key quantitative benchmarks across different cellular contexts, highlighting the variability that researchers must account for.
Table 1: Editing Efficiency and Kinetics Across Human Cell Types
| Cell Type | Example Locus | Avg. Indel Efficiency (%) | DSB Repair Half-Life (hr) | Error-Prone Repair Contribution | Primary Citation |
|---|---|---|---|---|---|
| K562 (Myelogenous Leukemia) | LBR | High (~80%) | Variable, up to ~10 hr | Significant | [97] |
| Embryonic Stem Cells (Mouse) | PigA | High (59-97% for exonic) | Not Measured | Large deletions >9.5 kb observed | [98] |
| Hematopoietic Stem Cells (Human) | BCL11A | High (Clinically effective) | Not Measured | Frequent kilobase-scale deletions | [14] |
| HEK293 (Human Embryonic Kidney) | Various | Typically High | Not Measured | Standard for benchmarking | [81] |
| Primary Cardiac Fibroblasts | N/A | Feasible for screening | Not Measured | Used in functional genomics | [99] |
Table 2: Impact of Genetic and Sequence Determinants on Editing Outcomes
| Determinant | Impact on Efficiency | Impact on Outcome Fidelity | Experimental Evidence |
|---|---|---|---|
| Protospacer Sequence | Strongly influences cutting rate (k~c~) | Dictates blunt vs. staggered break profile; affects single-nucleotide insertion precision [81] | BreakTag profiling of ~150,000 sites [81] |
| Human Genetic Variation | Can alter local sequence and accessibility | Impacts configuration of Cas9 cuts and DSB repair outcome; critical for personalized therapy [81] | Machine learning on BreakTag data [81] |
| DNA-PKcs Inhibition | Can enhance HDR efficiency in some contexts | Dramatically increases frequency of large deletions and chromosomal translocations [14] | CAST-Seq and LAM-HTGTS analysis [14] |
| p53 Status | TP53-knockout can increase proliferation post-editing | Associated with increased genome instability and large chromosomal aberrations [14] | Analysis in multiple human cell types [14] |
| Microhomology Regions | Not a direct efficiency factor | Strongly predisposes repair to MMEJ, leading to characteristic, predictable deletions [97] | Kinetics and sequencing of repair outcomes [97] |
Essential Methodologies for Comprehensive Efficacy Assessment
A critical lesson from recent research is that reliance on any single method, particularly short-range PCR amplification, can lead to a dramatic underestimation of unintended genetic damage [14] [98]. A robust assessment requires a multi-faceted approach.
BreakTag for Genome-Wide DSB Profiling
Principle: BreakTag is a versatile, next-generation sequencing-based method for profiling the genome-wide landscape of Cas9-induced DSBs, including their location, frequency, and end structures, at nucleotide resolution [81].
Detailed Protocol:
- In Vitro Digestion: Incubate purified genomic DNA (gDNA) with Cas9 ribonucleoprotein (RNP) complexes.
- End Repair & A-Tailing: Prepare the free DSB ends for ligation. This step also enriches for DSBs with single-stranded overhangs.
- Adapter Ligation: Ligate an adaptor containing a Unique Molecular Identifier (UMI) for absolute DSB counting and a sample barcode for multiplexing.
- Tagmentation: Use Tn5 transposase to fragment the DNA and add sequencing adapters in a single step.
- PCR Amplification: Amplify the ligated fragments. DSB enrichment occurs during this PCR step, resulting in a fast (<6 hours) and cost-efficient library.
- Sequencing & Analysis: Sequence the libraries and analyze data with the companion tool
BreakInspectoRto identify and count Cas9-induced DSBs.
Key Insight: BreakTag revealed that approximately 35% of SpCas9 DSBs are staggered (not blunt), a finding with major implications for predicting repair outcomes [81].
Measuring DSB Repair Kinetics at Single Loci
Principle: This method combines an inducible Cas9 system with time-series sampling and modeling to quantify the rates of cutting and repair for a specific genomic locus [97].
Detailed Protocol:
- Cell Line Engineering: Establish a clonal cell line (e.g., K562) with a stably integrated, inducible Cas9 construct. A common system uses a ligand-responsive destabilizing domain (e.g., Shield-1) to control Cas9 stability.
- sgRNA Transfection: Transiently transfect cells with a plasmid encoding a specific sgRNA.
- Induction and Time-Series Sampling: Stabilize Cas9 by adding Shield-1 to initiate synchronized cutting. Collect cell samples at multiple time points post-induction (e.g., from 0 to 60 hours).
- Amplicon Sequencing: For each time point, amplify a ~300-500 bp region surrounding the target site by PCR and perform high-throughput sequencing.
- Quantitative Modeling: Fit the kinetic model (Figure 1) to the time-series data of intact and indel allele fractions to estimate the rate constants k~c~, k~p~, and k~m~.
Key Insight: This approach demonstrated that repair is often slow (half-life up to ~10 hours) and that both classical and microhomology-mediated end joining contribute to erroneous repair, with their balance changing over time [97].
Detecting Large Structural Variations
Principle: Standard short-read amplicon sequencing can miss large deletions and complex rearrangements if primer binding sites are lost. Dedicated methods are required for their detection [14] [98].
Detailed Protocol (Long-Range PCR Genotyping):
- Long-Range PCR: Using polymerase kits designed for amplifying large fragments, perform PCR with primers placed several kilobases upstream and downstream of the target site.
- Gel Electrophoresis: Analyze PCR products by gel electrophoresis. Fragments smaller than the wild-type product indicate large deletions.
- Sequencing Verification: Purify aberrant bands and sequence them using Sanger sequencing or long-read sequencing technologies (e.g., PacBio) to characterize the exact nature of the rearrangement.
- Alternative Methods: Techniques like CAST-Seq or LAM-HTGTS are specifically designed to nominate and validate chromosomal translocations and other SVs genome-wide [14].
Key Insight: Studies using these methods have found that large deletions extending over many kilobases are a frequent outcome of Cas9 cutting in various cell types, including therapeutically relevant hematopoietic stem cells [98].
Figure 2. A Comprehensive Workflow for Assessing CRISPR-Cas9 Editing Efficacy. This integrated approach combines DSB mapping, kinetic analysis, and structural variation detection to build a complete picture of editing outcomes, balancing efficiency with safety.
The Scientist's Toolkit: Key Reagents and Solutions
Table 3: Essential Research Reagents for Editing Efficacy Analysis
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| High-Fidelity Cas9 | Reduces off-target cleavage while maintaining on-target activity. | Improving the specificity of editing in sensitive applications like therapeutic development [14]. |
| Inducible Cas9 System | Enables temporal control of Cas9 activity (e.g., via Shield-1 ligand). | Synchronizing DSB formation for accurate measurement of repair kinetics (k~c~, k~p~, k~m~) [97]. |
| BreakTag Kit | Provides a streamlined protocol for end-repair, adapter ligation, and library prep. | Unbiased, genome-wide mapping of Cas9 DSBs and their end structures (blunt vs. staggered) [81]. |
| Long-Range PCR Kit | Amplifies DNA fragments up to 20+ kb with high fidelity. | Detecting large deletions and genomic rearrangements missed by short-amplicon sequencing [98]. |
| DNA-PKcs Inhibitors (e.g., AZD7648) | Inhibits the classical NHEJ DNA repair pathway. | Experimentally shifting repair balance to study alternative pathways; caution required due to high risk of SVs [14]. |
BreakInspectoR Software |
Bioinformatics pipeline for identifying and counting Cas9-induced DSBs in BreakTag data. | Nominating and quantifying on- and off-target sites from BreakTag sequencing datasets [81]. |
Accurately benchmarking CRISPR-Cas9 efficacy requires moving beyond a single metric of indel percentage. A modern framework must integrate quantitative data on the kinetics of DSB repair, the spectrum of small indels, and the prevalence of large structural variations, all while acknowledging the profound influence of cellular context and genetic background. The methodologies detailed here—from BreakTag and kinetic modeling to long-range genotyping—provide the necessary toolkit for this comprehensive assessment.
The future of precision genome editing lies in leveraging these complex datasets to build predictive models. As research begins to link specific DSB profiles, such as staggered versus blunt ends, to predictable repair outcomes, the ability to design editing strategies with high precision and safety will be greatly enhanced [81]. For clinical translation, this rigorous, multi-faceted assessment is not merely a best practice but an essential requirement to ensure both the efficacy and safety of CRISPR-based therapies.
Targeted genomic manipulation represents one of the most transformative advancements in modern molecular biology, enabling researchers to precisely modify genetic sequences to elucidate gene function and develop novel therapeutic strategies. At the core of this technology lies the ability to programmably induce DNA double-strand breaks (DSBs) at specific genomic loci, thereby activating endogenous cellular repair mechanisms that can be harnessed to introduce desired genetic modifications [100] [101]. The evolution of genome editing platforms has progressed through multiple generations, from meganucleases and zinc-finger nucleases (ZFNs) to transcription activator-like effector nucleases (TALENs) and, most recently, the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system [101] [102]. Each technology operates on the fundamental principle of creating a targeted DSB, but differs significantly in its molecular architecture, DNA recognition mechanism, and practical implementation [103] [104]. This review provides a comprehensive technical comparison of these major genome editing platforms, with particular emphasis on their mechanisms for DSB formation, experimental parameters, and suitability for various research and therapeutic applications.
Fundamental Mechanisms of Programmable Nucleases
Architectural Principles and DNA Recognition
Programmable nucleases share a common functional architecture consisting of a DNA recognition module coupled to a nuclease domain, but implement this architecture through distinct molecular mechanisms.
ZFNs are chimeric proteins composed of a Cys2-His2 zinc-finger DNA-binding domain fused to the non-specific cleavage domain of the FokI restriction endonuclease [100] [104]. Each zinc-finger domain consists of approximately 30 amino acids arranged in a conserved ββα configuration that recognizes 3-4 base pairs (bp) in the major groove of DNA [100] [101]. Typically, 3-6 zinc-finger motifs are linked in tandem to recognize 9-18 bp sequences. ZFNs function as pairs, with each monomer binding opposite DNA strands and the FokI domains dimerizing to create a DSB in the spacer region (typically 5-6 bp) separating the two binding sites [101] [104].
TALENs similarly employ the FokI nuclease domain but utilize DNA-binding domains derived from transcription activator-like effectors (TALEs) from Xanthomonas bacteria [100] [101]. Each TALE repeat comprises 33-35 amino acids and recognizes a single bp through two hypervariable residues known as repeat-variable diresidues (RVDs). The RVD code enables predictable DNA recognition: NG recognizes T, NI recognizes A, HD recognizes C, and NN/HN/NK recognizes G [103] [101]. Like ZFNs, TALENs function as pairs with binding sites separated by a 12-19 bp spacer and require FokI dimerization for DSB formation [101].
CRISPR-Cas9 fundamentally differs from ZFNs and TALENs by utilizing an RNA-guided DNA recognition mechanism [2] [102]. The system consists of the Cas9 nuclease complexed with a guide RNA (gRNA) that includes a CRISPR RNA (crRNA) component with a 18-20 nucleotide spacer sequence complementary to the target DNA, and a trans-activating crRNA (tracrRNA) that facilitates complex formation [2] [105]. Cas9 recognizes target sequences adjacent to a protospacer adjacent motif (PAM; 5'-NGG-3' for Streptococcus pyogenes Cas9), with the gRNA-DNA complementarity directing target specificity [105] [102]. Upon target recognition, Cas9 undergoes conformational changes that activate its RuvC and HNH nuclease domains to cleave opposite DNA strands, creating a DSB 3 bp upstream of the PAM sequence [2] [102].
Table 1: Core Architecture and DNA Recognition Mechanisms
| Feature | ZFNs | TALENs | CRISPR-Cas9 |
|---|---|---|---|
| DNA recognition moiety | Zinc-finger proteins | TALE proteins | gRNA (crRNA:tracrRNA) |
| Recognition code | 3-4 bp per zinc finger | 1 bp per TALE repeat | RNA-DNA complementarity |
| Nuclease domain | FokI | FokI | Cas9 (RuvC & HNH domains) |
| Dimerization required | Yes | Yes | No |
| PAM/PAM-like requirement | No | Target must begin with 'T' | 5'-NGG-3' (for SpCas9) |
| Typical target length | 9-18 bp per monomer | 14-20 bp per monomer | 20 nt + PAM |
Double-Strand Break Formation and Characteristics
The molecular mechanisms of DSB formation significantly influence editing efficiency and outcome. ZFNs and TALENs both utilize the FokI nuclease domain, which must dimerize to become active, creating predominantly 5' overhangs of 4-5 nucleotides [101] [104]. This dimerization requirement enhances specificity by effectively doubling the recognition site length, but necessitates careful design to ensure proper spacing and orientation between binding sites.
In contrast, Cas9 functions as a single protein with two distinct nuclease domains: the HNH domain cleaves the DNA strand complementary to the gRNA, while the RuvC-like domain cleaves the non-complementary strand [2] [102]. This results in a blunt-ended DSB positioned 3 bp upstream of the PAM sequence [102]. The Cas9-gRNA complex identifies target sites through a combination of 3D and 1D diffusion, with PAM recognition triggering local DNA melting and R-loop formation through sequential gRNA-DNA pairing beginning at the PAM-proximal "seed" region [2].
The following diagram illustrates the fundamental mechanism of CRISPR-Cas9 induced DSB formation:
Comparative Performance Metrics
Efficiency, Specificity, and Practical Considerations
The practical implementation of genome editing technologies involves balancing multiple performance parameters, including editing efficiency, specificity, and practical workflow considerations.
Table 2: Performance and Practical Implementation Comparison
| Parameter | ZFNs | TALENs | CRISPR-Cas9 |
|---|---|---|---|
| Targeting efficiency | Variable (context-dependent) | High | Very high |
| Off-target effects | Lower than CRISPR-Cas9 [103] | Lower than CRISPR-Cas9 [103] | Higher (gRNA-dependent) [103] [102] |
| Design complexity | High (context-dependent recognition) [100] [104] | Medium (modular code) [100] | Very low (RNA-DNA complementarity) [103] [102] |
| Construction time | ~1 month (with optimization) [101] | ~1 month [101] | Within a week [101] |
| Cost | High [103] [101] | Medium [103] [101] | Low [103] [101] |
| Multiplexing capacity | Low | Low | High (multiple gRNAs) [105] |
| Delivery constraints | Moderate size | Large size challenging for viral delivery [101] | Cas9 size challenging for AAV delivery [101] |
CRISPR-Cas9 generally offers superior editing efficiency compared to ZFNs and TALENs, particularly for multiplexed applications where multiple gRNAs can be expressed simultaneously to target several genomic loci [105]. However, concerns regarding off-target effects remain more pronounced for CRISPR-Cas9 due to its tolerance of mismatches, particularly in the PAM-distal region of the target sequence [2] [102]. Both ZFNs and TALENs demonstrate higher specificity, attributed to their longer recognition sequences and, in the case of ZFNs, the requirement for obligate heterodimer formation in optimized designs [103] [104].
From a practical standpoint, CRISPR-Cas9 has democratized genome editing due to its simple design principles, rapid construction, and lower costs [103] [102]. The modularity of ZFNs is limited by context-dependent effects between adjacent zinc fingers, necessitating sophisticated optimization strategies [100] [104]. While TALENs employ a more straightforward recognition code, the highly repetitive nature of TALE arrays presents cloning challenges [100]. CRISPR-Cas9 bypasses these issues by relying on Watson-Crick base pairing for target recognition, enabling straightforward gRNA design against virtually any sequence adjacent to a PAM [105] [102].
DNA Repair Pathway Engagement
All three nuclease platforms induce DSBs that are processed by endogenous cellular repair mechanisms, primarily non-homologous end joining (NHEJ) and homology-directed repair (HDR) [100] [101]. The error-prone NHEJ pathway often results in small insertions or deletions (indels) that can disrupt gene function, while HDR enables precise genetic modifications using exogenous donor templates [101] [102].
Recent studies utilizing UMI-DSBseq for single-molecule quantification of repair dynamics have revealed that a substantial proportion of CRISPR-Cas9-induced DSBs are repaired precisely without mutagenic indels [6]. In tomato protoplasts, precise repair accounted for up to 70% of all repair events, with indel frequencies ranging between 15-41% despite cleavage rates of 64-88% across three targets [6]. The high fidelity of endogenous repair mechanisms thus significantly impacts the observed efficiency of mutagenesis, with precise repair competing with error-prone pathways.
The balance between NHEJ and HDR varies across cell types and is influenced by cell cycle stage, with HDR being most active in late S and G2 phases [2] [102]. While all nuclease platforms engage these repair pathways, the structure of the DSB ends may influence repair outcomes, with Cas9 generating blunt ends versus the overhangs created by FokI nucleases [2] [101].
Experimental Design and Workflow
Target Selection and Validation
Successful genome editing requires careful target selection and validation strategies tailored to each nuclease platform. For CRISPR-Cas9, target sites must be adjacent to PAM sequences (5'-NGG-3' for SpCas9) and should be selected to minimize off-target activity using available design tools [105]. The seed sequence (8-10 bases at the 3' end of the gRNA targeting sequence) requires perfect complementarity for efficient cleavage, while mismatches in the PAM-distal region are more tolerated [2] [105]. For ZFNs and TALENs, target sites must accommodate paired binding sites with appropriate spacing and orientation for FokI dimerization [101] [104].
Experimental validation should include comprehensive assessment of both on-target efficiency and off-target effects. For CRISPR-Cas9, this can be facilitated through the use of high-fidelity Cas9 variants (e.g., eSpCas9, SpCas9-HF1, HypaCas9) that reduce off-target cleavage while maintaining on-target activity [105]. The workflow for quantifying DSB formation and repair kinetics can be enhanced using molecular tools like UMI-DSBseq, which enables simultaneous quantification of DSB intermediates and repair products at single-molecule resolution [6].
The following diagram outlines a generalized experimental workflow for nuclease validation and DSB repair analysis:
Research Reagent Solutions
Table 3: Essential Research Reagents for DSB Formation Studies
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Nuclease Expression Systems | SpCas9, SaCas9, FnCas9 [101] | DSB induction; Variants offer different PAM specificities and sizes |
| Guide RNA Systems | sgRNA plasmids, crRNA:tracrRNA dual RNA [2] [105] | Target specification; Chemical modifications can enhance stability [2] |
| Delivery Tools | PEG-mediated RNP delivery [6], AAV, Lentivirus, Electroporation [104] | Introduction of editing components into cells; RNP delivery offers rapid activity [6] |
| Repair Pathway Modulators | NHEJ inhibitors, HDR enhancers | Influence repair outcome toward desired pathway |
| Detection & Validation Tools | UMI-DSBseq [6], T7E1 assay, NGS-based methods | Quantify DSB formation, repair kinetics, and editing outcomes |
| Specificity-Enhanced Variants | eSpCas9(1.1), SpCas9-HF1, HypaCas9 [105] | Reduce off-target effects while maintaining on-target activity |
| Nickase Variants | Cas9n (D10A) [105] | Generate single-strand breaks for enhanced specificity applications |
Applications and Future Perspectives
The applications of programmable nucleases span diverse fields including functional genomics, disease modeling, gene therapy, and agricultural biotechnology [102]. CRISPR-Cas9 has become the predominant platform for most research applications due to its simplicity and versatility [103] [102]. However, ZFNs and TALENs maintain important niches, particularly in therapeutic contexts where their higher specificity and longer development history offer advantages [103] [104]. Additionally, TALENs provide unique capabilities for mitochondrial genome editing (mito-TALENs), where RNA import challenges limit CRISPR applications [103].
Recent advancements have focused on addressing limitations of each platform. For CRISPR-Cas9, this includes developing high-fidelity variants, PAM-flexible enzymes (e.g., xCas9, SpCas9-NG, SpRY), and base editors that enable precise nucleotide changes without DSBs [106] [105]. For ZFNs and TALENs, improvements have centered on enhanced DNA-binding specificity and optimized delivery methods [104].
The field continues to evolve with emerging technologies such as prime editing, CRISPR-associated recombinases, and epigenetic editors expanding the genome editing toolbox [106]. Nevertheless, the fundamental principles of DSB formation and repair elucidated through comparative studies of ZFNs, TALENs, and CRISPR-Cas9 continue to inform the development of next-generation genome editing platforms with enhanced precision and safety profiles for research and therapeutic applications.
The advent of RNA-guided CRISPR-associated transposase (CAST) systems marks a paradigm shift in genome engineering, offering a powerful alternative to traditional CRISPR-Cas9 mechanisms that rely on double-strand break (DSB) formation and endogenous DNA repair pathways. These systems, which include both Cascade-based (Type I-F) and single-effector (Type V-K) architectures, enable programmable, site-specific integration of large DNA payloads without creating DSBs. This technical review evaluates CAST molecular mechanisms, quantifies current performance across prokaryotic and eukaryotic systems, and provides detailed experimental frameworks for implementing these technologies in research and therapeutic development. As the field advances toward clinical application, CAST systems address critical limitations of DSB-dependent editing—including indel formation, repair pathway dependence, and cytotoxicity—while introducing new considerations for specificity and efficiency optimization.
The CRISPR-Cas9 system has revolutionized genome engineering by providing unprecedented precision in creating targeted double-strand breaks. This mechanism relies on two key components: a guide RNA (gRNA) that specifies the genomic target, and the Cas9 endonuclease that creates the DSB approximately 3-4 nucleotides upstream of the protospacer adjacent motif (PAM) sequence [105]. Cellular repair of these breaks occurs primarily through two pathways: error-prone non-homologous end joining (NHEJ), which often results in insertions or deletions (indels) that disrupt gene function, or homology-directed repair (HDR), which enables precise edits using a DNA template [105] [28].
While highly effective, this DSB-dependent mechanism presents significant limitations for therapeutic applications. NHEJ can produce unpredictable indels, while HDR efficiency is constrained by cell cycle phase and varies considerably between cell types [30]. In nondividing cells such as neurons and cardiomyocytes, DSB repair pathways differ dramatically from dividing cells, resulting in extended repair timelines and altered editing outcomes [39]. Furthermore, the integration of large DNA sequences via HDR remains inefficient, limiting applications that require insertion of therapeutic transgenes.
CAST systems represent a distinct evolutionary adaptation where transposons have co-opted CRISPR-Cas systems for RNA-guided integration. Unlike standard CRISPR-Cas systems, CAST systems direct the integration of large DNA fragments without creating double-strand breaks, instead leveraging transposition machinery for precise insertion [107] [30]. This mechanism operates independently of host repair pathways, potentially enabling more predictable editing outcomes across diverse cell types.
Molecular Architecture and Mechanisms
CAST systems comprise two functional modules: a targeting complex for RNA-guided DNA recognition and a transposase complex for DNA integration. The specific architecture varies between system subtypes, with Type I-F and Type V-K representing the most well-characterized classes.
Type I-F Cascade CAST Systems
Type I-F systems employ a multi-protein Cascade (CRISPR-associated complex for antiviral defense) complex for target recognition:
- Targeting Complex: Cas6, Cas7, and Cas8 proteins form the Cascade complex with crRNA for programmable DNA targeting [30] [108].
- Transposase Complex: TnsA (endonuclease), TnsB (DDE-transposase), TnsC (AAA+ ATPase adaptor), and TniQ (targeting module adaptor) [30].
- Mechanism: Type I-F systems utilize a "cut-and-paste" transposition mechanism where TnsA and TnsB form a heteromeric transposase that excises the donor DNA before integration [30].
The following diagram illustrates the molecular architecture and integration mechanism of Type I-F Cascade CAST systems:
Type V-K CAST Systems
Type V-K systems utilize a single effector protein for target recognition, offering a more compact architecture:
- Targeting Complex: Cas12k effector protein with guide RNA [30] [108].
- Transposase Complex: TnsB (DDE-transposase), TnsC (AAA+ ATPase adaptor), and TniQ (targeting module adaptor) – but lacks TnsA [30] [108].
- Mechanism: Without TnsA for second-strand cleavage, Type V-K systems typically generate cointegrate products through a replicative transposition pathway [30] [108].
The following diagram illustrates the molecular architecture and integration mechanism of Type V-K CAST systems:
Comparative Performance Analysis
CAST systems demonstrate distinct performance characteristics across prokaryotic and eukaryotic environments, with efficiency varying significantly based on system architecture, delivery method, and target organism.
Table 1: Performance Characteristics of CAST Systems in Prokaryotic Systems
| System Type | Organism | Insert Size | Efficiency | Integration Specificity | Key Features |
|---|---|---|---|---|---|
| Type I-F (VchCAST) | E. coli | Up to 10 kb | ~100% [109] | >99% on-target [109] | Single-plasmid system (pSPIN), marker-free |
| Type I-F (VchCAST) | E. coli | ~15.4 kb | High efficiency [30] | High specificity [30] | Cut-and-paste mechanism |
| Type V-K (ShCAST) | E. coli | Up to 30 kb [30] | Moderate efficiency [30] | 40-60% off-targets [108] | Replicative transposition |
| Type V-K (MG64-1) | E. coli | Not specified | Up to 80% [108] | <7% off-targets [108] | Metagenomically discovered |
Table 2: Performance Characteristics of CAST Systems in Eukaryotic Systems
| System Type | Cell Line | Insert Size | Efficiency | Key Features |
|---|---|---|---|---|
| Type I-F CAST | HEK293 | ~1.3 kb | ~1% [30] | Requires bacterial chaperone ClpX |
| Type V-K CAST (engineered) | HEK293T | 2.6 kb | 0.06% [30] | Plasmid target |
| Type V-K (MG64-1) | HEK293 | 3.2 kb | ~3% [30] [108] | AAVS1 safe harbor locus |
| Type V-K (MG64-1) | K562 | 3.6 kb | ~3% [30] [108] | Therapeutic donor |
| Type V-K (MG64-1) | Hep3B | 3.6 kb | <0.05% [30] [108] | Therapeutic donor |
Experimental Framework for CAST Implementation
Implementing CAST systems requires careful consideration of vector design, component delivery, and validation methods. The following section outlines established protocols for both prokaryotic and eukaryotic systems.
Prokaryotic Implementation (INTEGRATE System)
The optimized INTEGRATE system employs a single-plasmid design (pSPIN) for high-efficiency integration in E. coli:
Vector Design and Components:
- pSPIN Plasmid: Single plasmid encoding crRNA, Cas proteins, and transposase machinery upstream of the mini-transposon donor DNA [109].
- Promoter Selection: Strong constitutive promoters (e.g., J23119) drive higher integration efficiency, though even weak promoters (J23114) achieve ~90% efficiency at room temperature [109].
- Donor Design: Mini-transposon flanked by terminal inverted repeats (TIRs) recognized by TnsB [109].
Experimental Protocol:
- Cloning: Insert target-specific spacer into CRISPR array and desired payload into mini-transposon region [109].
- Transformation: Introduce pSPIN plasmid into electrocompetent E. coli BL21(DE3) or other strains [109].
- Cultivation: Grow transformed cells at 30°C or room temperature to maximize integration efficiency [109].
- Validation: Screen colonies via PCR and sequence integration junctions to verify precise insertion ~50 bp downstream of target site [109].
Efficiency Optimization:
- Temperature Modulation: Growth at 30°C or room temperature significantly increases efficiency compared to 37°C [109].
- Promoter Strength: Stronger constitutive promoters correlate with higher integration rates [109].
- Multiplexing: Co-expression of multiple guide RNAs enables simultaneous integration at 2-3 genomic loci with 50-80% efficiency per site [109].
Eukaryotic Implementation (Type V-K CAST Systems)
Implementation in human cells requires additional engineering for nuclear localization and function:
System Engineering:
- Nuclear Localization: Addition of nuclear localization signals (NLS) to all protein components [108].
- Host Factor Co-expression: Co-expression of prokaryotic ribosomal protein S15 enhances integration efficiency [30].
- Donor Design: Linear donor fragments with defined ends minimize co-integrate formation [108].
Delivery Methods:
- Plasmid Transfection: Multiple plasmids encoding CAST components and donor DNA [108].
- Virus-Like Particles (VLPs): Engineered VLPs pseudotyped with VSVG/BRL envelopes achieve >97% transduction efficiency in human iPSC-derived neurons [39].
Validation and Specificity Assessment:
- On-Target Verification: PCR amplification across integration junctions with sequencing confirmation [108].
- Off-Target Profiling: Whole-genome sequencing (Tn-seq) to identify rare off-target events [109] [108].
- Functional Assays: Expression analysis of integrated transgenes (e.g., Factor IX at safe harbor loci) [108].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of CAST systems requires specific molecular tools and reagents. The following table catalogues essential components for establishing CAST-based genome engineering.
Table 3: Essential Research Reagents for CAST System Implementation
| Reagent Category | Specific Examples | Function | Implementation Notes |
|---|---|---|---|
| Targeting Components | Cas12k (Type V-K), Cascade complex (Type I-F) | RNA-guided DNA recognition | Nuclear localization signals (NLS) required for eukaryotic use |
| Transposase Components | TnsB, TnsC, TniQ, TnsA (Type I-F only) | Donor DNA excision and integration | TnsA presence determines cut-and-paste vs. replicative mechanism |
| Guide RNA | crRNA with specific repeat structures, tracrRNA | System targeting specificity | Conserved stem-loop structures in tracrRNA critical for function |
| Donor Template | Mini-transposon with Terminal Inverted Repeats (TIRs) | Genetic cargo for integration | Linear donors reduce co-integrate formation in Type V-K systems |
| Vector Systems | pSPIN (single-plasmid INTEGRATE) | Component delivery | All-in-one design dramatically improves efficiency |
| Host Factors | Ribosomal protein S15, ClpX (eukaryotic systems) | Enhance integration efficiency | Prokaryotic factors required for function in eukaryotic environment |
| Delivery Tools | VSVG/BRL-pseudotyped VLPs, electroporation | Component delivery to cells | VLPs effective for difficult-to-transfect cells like neurons |
CAST systems represent a significant advancement beyond DSB-dependent CRISPR-Cas9 genome editing, offering a programmable platform for precise DNA integration without relying on endogenous repair mechanisms. While current eukaryotic efficiency remains lower than prokaryotic applications, recent engineering advances—including metagenomic discovery of novel systems (MG64-1), optimization of delivery methods, and host factor identification—are rapidly addressing these limitations.
The unique capabilities of CAST systems for kilobase-scale integration position them as transformative tools for therapeutic development, particularly for diseases requiring insertion of large therapeutic transgenes. As specificity and efficiency continue to improve through protein engineering and mechanistic understanding, CAST technologies are poised to become indispensable components of the genome engineering toolkit, working alongside rather than replacing existing CRISPR-based approaches to enable previously impossible genetic modifications.
The translation of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 from a revolutionary laboratory tool to a clinical therapeutic has marked a new era in genetic medicine. As the first approved CRISPR-based therapies are administered to patients and numerous clinical trials progress, the landscape is characterized by promising efficacy data and an increasing focus on comprehensive safety profiles. This whitepaper synthesizes the most current safety and efficacy data from recent human studies, framing these clinical findings within the fundamental context of CRISPR-Cas9's mechanism for double-strand break (DSB) formation and repair. For researchers, scientists, and drug development professionals, understanding this intricate relationship between molecular mechanism and clinical outcome is paramount for designing safer, more effective next-generation therapies.
Current Clinical Trial Landscape and Efficacy Outcomes
The clinical application of CRISPR-Cas9 has expanded rapidly, with over 100 clinical trials currently underway [14]. The following analysis summarizes efficacy data from key late-stage trials across diverse disease areas, illustrating the potent therapeutic potential of genome editing.
Table 1: Efficacy Data from Recent CRISPR-Cas9 Clinical Trials
| Therapeutic / Trial Identifier | Target / Condition | Delivery Method | Key Efficacy Findings | Reference / Phase |
|---|---|---|---|---|
| Casgevy (exa-cel) | BCL11A / Sickle Cell Disease & β-thalassemia | Ex Vivo | Sustained increase in fetal hemoglobin; freedom from vaso-occlusive crises in SCD; transfusion independence in TBT | Approved Therapy [8] |
| NTLA-2001 (Intellia) | TTR / hATTR Amyloidosis | LNP, in vivo IV | ~90% sustained reduction in serum TTR protein over 24 months; disease stability or improvement | Phase 1 [8] |
| NTLA-2002 (Intellia) | KLKB1 / Hereditary Angioedema (HAE) | LNP, in vivo IV | 86% avg. reduction in kallikrein; 8 of 11 high-dose participants attack-free over 16 weeks | Phase 1/2 [8] |
| CTX310 | ANGPTL3 / Dyslipidemias | LNP, in vivo IV | ~50% reduction in LDL cholesterol; ~55% reduction in triglycerides; effects sustained ≥60 days | Phase 1 [110] |
| BRL-201 | PD-1 / Lymphoma | Ex Vivo (non-viral) | Sustained remission, patient cancer-free for over 5 years | Phase 1/2 [111] |
The data reveal several key trends. First, high efficacy rates are consistently achieved, with protein knockdown levels often exceeding 85-90% for in vivo liver-targeted therapies [8] [110]. Second, the effects are durable, with sustained biomarker correction and clinical benefit observed over multiple years in the longest-running trials [8] [111]. Finally, the successful application of editing across diverse delivery methods—from complex ex vivo stem cell engineering to systemic in vivo administration—highlights the maturing versatility of the platform.
Novel Workflows and Personalized Approaches
A landmark case in 2025 demonstrated the potential for ultra-personalized CRISPR medicine. Researchers developed a bespoke in vivo therapy for an infant with a rare, lethal genetic disease, CPS1 deficiency [8]. The workflow, summarized in the diagram below, was executed in a remarkably short timeframe of just six months from development to delivery, establishing a regulatory precedent for rapid, patient-specific therapies.
This case also provided critical proof-of-concept for LNP-mediated redosing. The patient safely received multiple doses, which incrementally increased the percentage of edited cells and improved clinical symptoms [8]. This challenges the long-held paradigm that genome editing is a strictly one-time intervention and opens new avenues for dose titration.
Safety Profile: From On-Target Edits to Structural Variations
While efficacy is robust, the safety profile of CRISPR-Cas9 therapies is complex and intimately tied to the fundamental mechanism of DSB formation and repair. Safety assessments have evolved beyond initial concerns about off-target effects to include a broader spectrum of genomic alterations.
Table 2: Summary of Key Safety Considerations in CRISPR-Cas9 Clinical Trials
| Safety Concern | Description | Influencing Factors | Detection Methods |
|---|---|---|---|
| On-Target Structural Variations | Large deletions (kb-Mb scale), chromosomal rearrangements, loss of heterozygosity. | Use of DNA-PKcs inhibitors; target locus; cell type. | CAST-Seq, LAM-HTGTS, single-cell sequencing [14]. |
| Off-Target Editing | Unintended edits at genomic sites with sequence similarity to the target. | gRNA design, Cas9 variant specificity, delivery method. | GUIDE-seq, Digenome-seq, computational prediction [112]. |
| On-Target Genomic Toxicity | Deletion of critical cis-regulatory elements during editing. | Target site within the genome; size of deletion. | Long-read sequencing, functional assays [14]. |
| Immunogenic Responses | Immune reaction to bacterial-derived Cas protein or delivery vehicle. | Delivery method (viral vs. LNP), pre-existing immunity. | Immunoassays, monitoring of infusion reactions [8] [59]. |
The Central Role of DNA Repair and Structural Variations
The primary safety concern is the generation of double-strand breaks and their subsequent repair. While non-homologous end joining (NHEJ) or homology-directed repair (HDR) can yield desired edits, they can also lead to significant unintended consequences [113] [59]. Recent studies using advanced detection technologies have revealed that CRISPR-Cas9 editing, particularly under conditions that manipulate DNA repair pathways, can induce large structural variations (SVs), including:
- Kilobase- to megabase-scale deletions at the on-target site [14] [110]
- Chromosomal translocations between the target site and off-target sites [14] [59]
- Chromosomal losses and truncations [14]
A critical finding is that strategies to enhance HDR efficiency, such as using DNA-PKcs inhibitors (e.g., AZD7648), can dramatically increase the frequency of these SVs. One study reported a thousand-fold increase in chromosomal translocation frequency with such inhibitors [14]. Furthermore, these large deletions can confound standard PCR-based quality control methods if primer binding sites are deleted, leading to an overestimation of precise HDR rates [14]. The workflow for comprehensive safety assessment is evolving to incorporate these findings, as shown below.
The presence of such SVs in clinically relevant cells is a major focus. For example, kilobase-scale deletions have been observed at the BCL11A locus in hematopoietic stem cells (HSCs) edited for Casgevy production [14]. Given BCL11A's role in lymphoid development and cellular senescence, the long-term functional impact of these alterations requires careful monitoring [14].
Advanced Methodologies for Safety and Efficacy Assessment
Robust experimental protocols are essential to accurately characterize the safety and efficacy of CRISPR-based interventions. The following section details key methodologies cited in recent literature.
Protocol 1: Comprehensive Analysis of Editing Outcomes via Single-Cell DNA Sequencing
Background: Bulk sequencing methods can miss complex heterogenous editing outcomes, such as mixed genotypes and large SVs, leading to an incomplete safety profile [114] [14].
Workflow:
- Cell Preparation: Single-cell suspensions are generated from the edited cell population (e.g., clinically manufactured CAR-T cells or HSPCs).
- Microfluidic Partitioning: Cells are partitioned into nanoliter-scale reaction chambers using platforms like the Mission Bio Tapestri. Each chamber contains a single cell and a unique barcode.
- Cell Lysis and DNA Amplification: Cells are lysed within the chambers, and the genomic DNA is amplified. Barcodes are attached to all amplicons from a single cell.
- Targeted Sequencing: Pre-designed panels are used to amplify over 100 target loci simultaneously, including the primary on-target site and potential off-target sites identified in silico or empirically.
- Data Analysis: Bioinformatic pipelines deconvolute the barcoded sequences to reconstruct the complete genotype of each individual cell. This allows for the direct assessment of:
- Co-occurrence of edits (e.g., on-target and off-target edits in the same cell).
- Zygosity of edits (heterozygous vs. homozygous).
- Presence of large structural variations.
- Clonality and diversity of the edited cell population.
Significance: This protocol, as applied in [114], revealed a "unique editing pattern in nearly every edited cell," highlighting the critical need for single-cell resolution to fully understand the genetic landscape of a therapeutic cell product and ensure its safety.
Protocol 2: Genome-Wide Identification of Structural Variations
Background: Standard amplicon-based sequencing is inadequate for detecting large-scale deletions or translocations that extend beyond the PCR amplification site.
Workflow (CAST-Seq):
- DNA Extraction and Shearing: High-molecular-weight genomic DNA is extracted from edited cells and randomly sheared.
- Circularization: DNA fragments are circularized using DNA ligase under dilute conditions that favor self-ligation.
- Nested PCR: Two rounds of PCR amplification are performed using primers specific to the target genomic region(s) of interest (e.g., the CRISPR cut site). This selectively amplifies fusion fragments derived from chromosomal rearrangements.
- Library Prep and NGS: The PCR products are prepared for next-generation sequencing (NGS).
- Bioinformatic Analysis: Sequenced reads are mapped to the reference genome. Chimeric reads that map to two distinct genomic loci are identified as potential translocation or rearrangement events. The frequency and type of SVs are quantified.
Significance: CAST-Seq and related methods (LAM-HTGTS) are becoming essential tools for the sensitive detection of genotoxic SVs, as reported in [14]. These methods are crucial for pre-clinical safety assessment and for monitoring the genomic integrity of clinical products.
The Scientist's Toolkit: Key Research Reagent Solutions
Advancements in CRISPR clinical translation are supported by a growing toolkit of optimized research reagents. The following table details key materials used in the featured experiments and the broader field.
Table 3: Essential Research Reagents for CRISPR-Cas9 Therapy Development
| Reagent / Solution | Function | Application Context |
|---|---|---|
| Alt-R HDR Enhancer Protein | Boosts homology-directed repair efficiency up to two-fold in hard-to-edit cells like iPSCs and HSPCs. | Used for improving precise gene correction in ex vivo therapies. Compatible with multiple Cas systems [111]. |
| HiFi Cas9 Variants | Engineered SpCas9 proteins with enhanced specificity, reducing off-target editing while maintaining high on-target activity. | Critical for in vivo therapeutic applications where off-target effects are a major safety concern [112]. |
| Lipid Nanoparticles (LNPs) | Non-viral delivery vehicles for in vivo CRISPR component delivery (e.g., Cas9-gRNA RNP or mRNA). Naturally target liver cells. | Used in multiple clinical trials for liver-targeted diseases (e.g., hATTR, HAE, dyslipidemias). Enable potential redosing [8]. |
| Anti-CRISPR Proteins | Natural inhibitors of Cas proteins that can be used to rapidly turn off CRISPR activity post-editing, limiting exposure time. | Mitigates off-target effects; allows for temporal control of the editing process in ex vivo and in vivo settings [112]. |
| Mission Bio Tapestri Platform | A single-cell DNA sequencing platform for comprehensive genotyping of edited cell populations at multiple loci. | Used for deep safety profiling of clinical-grade cell products to identify complex editing outcomes and SVs [114]. |
The current clinical trial landscape for CRISPR-Cas9 demonstrates a technology transitioning from proof-of-concept to tangible therapeutic impact. Robust efficacy data across a range of diseases, particularly those amenable to liver editing and ex vivo hematopoietic stem cell modification, is highly promising. However, the evolving understanding of safety, particularly the risk of on-target structural variations linked to the core mechanism of DSB repair, underscores that efficacy and safety are two sides of the same coin. The future of the field hinges on the continued development of more precise editing tools, the integration of advanced detection methodologies like single-cell sequencing into standard workflows, and the thoughtful application of DNA repair pathway modulators. For drug development professionals, a deep mechanistic understanding of DSB formation and repair is no longer merely academic—it is an essential prerequisite for designing the next generation of safer, more effective CRISPR-based gene therapies.
Conclusion
The formation of double-strand breaks by CRISPR-Cas9 represents a transformative technology that has revolutionized genetic engineering and therapeutic development. Understanding the complete mechanistic pathway—from programmable DNA recognition through precise cleavage to cellular repair—provides the foundation for innovative applications across biomedical research and clinical medicine. While significant challenges remain in delivery optimization, off-target minimization, and repair pathway control, emerging technologies like base editing, prime editing, and CRISPR-associated transposons offer promising avenues to overcome these limitations. The successful clinical translation of CRISPR therapies for genetic disorders demonstrates tangible progress, yet future advancements will require interdisciplinary approaches combining structural biology, computational design, and delivery engineering. As validation methods improve and comparative analyses refine our understanding of genome-editing platforms, CRISPR technology is poised to enable increasingly precise interventions for complex diseases, ultimately fulfilling its potential to redefine therapeutic paradigms across medicine.
References
- 1. of Structures Endonucleases Reveal Cas -Mediated... 9 RNA [https://pmc.ncbi.nlm.nih.gov/articles/PMC4184034/]
- 2. DNA repair pathway choices in CRISPR-Cas9 mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8187289/]
- 3. A Guide to Efficient CRISPR gRNA Design: Principles and ... [https://www.genscript.com/a-guide-to-efficient-crispr-grna-design-principles-and-design-tools.html]
- 4. How to Design Your gRNA for CRISPR Genome Editing [https://blog.addgene.org/how-to-design-your-grna-for-crispr-genome-editing]
- 5. CRISPR: Guide to gRNA design [https://www.snapgene.com/guides/design-grna-for-crispr]
- 6. Uncovering the dynamics of precise repair at CRISPR ... [https://www.nature.com/articles/s41467-024-49410-x]
- 7. AI-powered CRISPR could lead to faster gene therapies ... [https://med.stanford.edu/news/all-news/2025/09/ai-crispr-gene-therapy.html]
- 8. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 9. Importance of the PAM Sequence in CRISPR Experiments [https://www.synthego.com/guide/how-to-use-crispr/pam-sequence/]
- 10. PAM identification by CRISPR-Cas effector complexes [https://pmc.ncbi.nlm.nih.gov/articles/PMC6546366/]
- 11. What Is the PAM Sequence for CRISPR and Where Is It? | IDT [https://www.idtdna.com/pages/support/faqs/what-is-a-pam-sequence-and-where-is-it-located]
- 12. Unraveling the mechanisms of PAMless DNA interrogation ... [https://www.nature.com/articles/s41467-024-47830-3]
- 13. Off-target effects in CRISPR/Cas9 gene editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10034092/]
- 14. The hidden risks of CRISPR/Cas: structural variations and ... [https://www.nature.com/articles/s41467-025-62606-z]
- 15. Overview CRISPR Clinical Trials 2025 - Learn | Innovate [https://crisprmedicinenews.com/clinical-trials/]
- 16. Rapid two-step target capture ensures efficient CRISPR- ... [https://www.sciencedirect.com/science/article/pii/S1097276525003016]
- 17. Exploring CRISPR-Cas: The transformative impact of gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506487/]
- 18. Structure basis for directional R-loop formation and substrate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5841471/]
- 19. Dynamic basis of supercoiling-dependent DNA ... [https://www.nature.com/articles/s41467-025-57703-y]
- 20. Article Cas12a domain flexibility guides R-loop formation ... [https://www.sciencedirect.com/science/article/pii/S1097276524004854]
- 21. Influence of PARP1 on CRISPR/Cas9 induced Double ... [https://www.sciencedirect.com/science/article/pii/S2001037025004039]
- 22. Cut Site Selection by the Two Nuclease Domains of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4036338/]
- 23. Frontiers | CRISPR /Cas9 technology in tumor research and drug ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1552741/full]
- 24. HNH proteins are a widespread component of phage DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4000778/]
- 25. Escherichia coli RuvC protein is an endonuclease that ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC453191/]
- 26. RuvC uses dynamic probing of the Holliday junction to ... [https://www.nature.com/articles/s41467-019-11900-8]
- 27. Weaponizing CRISPR/Cas9 for selective elimination of ... [https://www.sciencedirect.com/science/article/pii/S1568786425000369]
- 28. Revolution of Biotechnology with CRISPR - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322092/]
- 29. -Cas9: Shaping the Future of Targeted CRISPR Drug Development [https://www.news-medical.net/life-sciences/CRISPR-Cas9-Shaping-the-Future-of-Targeted-Drug-Development.aspx]
- 30. Advances in large-scale DNA engineering with the ... [https://www.nature.com/articles/s12276-025-01530-0]
- 31. Top scientific discoveries and breakthroughs for | CAS [https://www.cas.org/resources/cas-insights/scientific-breakthroughs-2025-emerging-trends-watch]
- 32. Dynamics and Mechanisms of CRISPR-Cas9 through ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9398989/]
- 33. Conformational control of DNA target cleavage by CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4859810/]
- 34. Quantitative assessment of engineered Cas9 variants for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8565309/]
- 35. Structural insights into DNA cleavage activation of CRISPR ... [https://www.nature.com/articles/s41467-017-01496-2]
- 36. The Conformational Dynamics of Cas9 Governing DNA ... [https://www.sciencedirect.com/science/article/pii/S2211124717318727]
- 37. HDR vs. NHEJ: CRISPR Gene Editing Techniques [https://www.zeclinics.com/blog/hdr-vs-nhej-in-crispr-gene]
- 38. HDR vs NHEJ: DNA Repair Pathway Comparison [https://invivobiosystems.com/crispr/hdr-vs-nhej/]
- 39. Characterizing and controlling CRISPR repair outcomes in ... [https://www.nature.com/articles/s41467-025-66058-3]
- 40. First CRISPR Therapies Approved for Sickle Cell Disease ... [https://www.goldbio.com/blogs/articles/crispr-therapies-sickle-cell-disease-thalassemia?srsltid=AfmBOor7eTrKy3MuQLkUKYV18BRpfW-ktcQ0d57nyJPbuLL_iGH3OOhx]
- 41. Revolutionary breakthrough: FDA CASGEVY, the first... approves [https://pmc.ncbi.nlm.nih.gov/articles/PMC11305803/]
- 42. First Gene FDA to Treat Patients with Approves ... Therapies Sickle [https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease]
- 43. First FDA Gene Editing Treatment for Approves ... CRISPR Sickle Cell [https://www.scientificamerican.com/article/fda-approves-first-crispr-gene-editing-treatment-for-sickle-cell-disease/]
- 44. Gene Therapy Leads to Improved Quality of Life in Patients ... [https://www.hematology.org/newsroom/press-releases/2025/gene-therapy-leads-to-improved-quality-of-life]
- 45. New treatment for sickle : cell pipeline highlights disease 2025 [https://www.labiotech.eu/in-depth/sickle-cell-disease-treatment/]
- 46. World's First Patient Treated with Personalized CRISPR ... [https://www.chop.edu/news/worlds-first-patient-treated-personalized-crispr-gene-editing-therapy-childrens-hospital]
- 47. Critical Quality Attributes of Lipid Nanoparticles and In Vivo ... [https://www.sciencedirect.com/science/article/pii/S1818087625000765]
- 48. Boosting RNA nanotherapeutics with V-ATPase activating ... [https://www.nature.com/articles/s41467-025-61688-z]
- 49. Design Strategies for Novel Lipid Nanoparticle for mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12497691/]
- 50. Advances in Antibody-Targeted Lipid Nanoparticles (Ab- ... [https://www.biochempeg.com/article/449.html]
- 51. CRISPR's Efficiency Triples With Spherical Nucleic Acid ... [https://news.feinberg.northwestern.edu/2025/09/05/crisprs-efficiency-triples-with-spherical-nucleic-acid-delivery-system/]
- 52. Lipid nanoparticle delivery of the CRISPR/Cas9 system ... [https://www.nature.com/articles/s41598-025-03671-8]
- 53. The hidden risks of CRISPR/Cas: structural variations and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12325979/]
- 54. Revolution of Biotechnology with CRISPR [https://www.nature.com/articles/s12276-025-01452-x]
- 55. CRISPR/Cas9 gene editing: A new hope for transthyretin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9574705/]
- 56. Early Findings from Novel CRISPR Therapy Show Promise ... [https://arci.org/nobel-prize-winning-science-being-applied-in-attr-amyloidosis/]
- 57. CRISPR-Based Therapy for Hereditary Angioedema [https://pubmed.ncbi.nlm.nih.gov/39445704/]
- 58. Pipeline [https://crisprtx.com/pipeline]
- 59. Challenges and Opportunities in the Application of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503735/]
- 60. Phage engineering and phage‐assisted CRISPR‐Cas ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10013820/]
- 61. Current Applications and the Future of Phage Therapy for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12189447/]
- 62. Global chromosome rearrangement induced by CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8989517/]
- 63. CMN Weekly (10 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-10-october-2025-your-weekly-crispr-medicine-news/]
- 64. Off-target effects in CRISPR-Cas genome editing for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12329535/]
- 65. Off-target effects in CRISPR-Cas genome editing for ... [https://www.sciencedirect.com/science/article/pii/S2162253125001908]
- 66. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 67. CRISPR 101: Off-Target Effects [https://blog.addgene.org/crispr-101-off-target-effects]
- 68. CRISPR-Cas9-mediated homology-directed repair for ... [https://www.sciencedirect.com/science/article/pii/S2162253124002312]
- 69. Comprehensive analysis of off-target and on-target effects ... [https://www.sciencedirect.com/science/article/pii/S2329050124001815]
- 70. Recent Updates of the CRISPR/Cas9 Genome Editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11164216/]
- 71. Delivery of Tissue-Targeted Scalpels: Opportunities and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7996671/]
- 72. Overcoming the Delivery Challenges in CRISPR/Cas9 ... [https://www.medsci.org/v22p3625.htm]
- 73. CRISPR/Cas9 systems: Delivery technologies and ... [https://www.sciencedirect.com/science/article/pii/S1818087623000818]
- 74. CRISPR Delivery Systems for Organ-Specific Targeting [https://www.sciencedirect.com/science/article/pii/S2950482125000336]
- 75. CRISPR-StAR enables high-resolution genetic screening ... [https://www.nature.com/articles/s41587-024-02512-9]
- 76. CRISPR-Cas9-mediated homology-directed repair for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11531618/]
- 77. Systematic quantification of HDR and NHEJ reveals effects ... [https://www.nature.com/articles/srep23549]
- 78. Methods for Optimizing CRISPR-Cas9 Genome Editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4976696/]
- 79. Frontiers | Mapping the therapeutic landscape of CRISPR - Cas for... 9 [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1558432/full]
- 80. Comprehensive analysis and accurate quantification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9586483/]
- 81. Linking CRISPR–Cas9 double-strand break profiles to ... [https://www.nature.com/articles/s41587-024-02238-8]
- 82. HDR knock in Templates [https://horizondiscovery.com/en/gene-editing/gene-editing-reagents/hdr-knock-in-templates]
- 83. Harnessing DSB repair to promote efficient homology- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8948305/]
- 84. Current Status and Challenges of DNA Base Editing Tools [https://pmc.ncbi.nlm.nih.gov/articles/PMC7474268/]
- 85. Back to Basics - Base & Prime Editing [https://frontlinegenomics.com/back-to-basics-base-and-prime-editing/]
- 86. Emerging trends in prime editing for precision genome ... [https://www.nature.com/articles/s12276-025-01463-8]
- 87. Elucidating the genetic mechanisms governing cytosine ... [https://www.nature.com/articles/s41467-025-59948-z]
- 88. Beyond the Double-Stranded Cut: Evolution of CRISPR ... [https://www.synthego.com/blog/beyond-crispr-methods/]
- 89. In vivo precision base editing to rescue mouse models of ... [https://www.sciencedirect.com/science/article/pii/S2162253125001763]
- 90. Search-and-replace genome editing without double-strand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6907074/]
- 91. Methods for detecting off-target effects of CRISPR/Cas9 [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002368]
- 92. Off-target interactions in the CRISPR-Cas9 Machinery [https://pubmed.ncbi.nlm.nih.gov/40688512/]
- 93. BLOG: Selecting the Right Gene Editing Off-Target Assay [https://seqwell.com/guide-to-selecting-right-gene-editing-off-target-assay/]
- 94. A versatile CRISPR/Cas9 system off-target prediction tool ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12144082/]
- 95. CRISPR Genome Editing & NGS | Off-target analysis and ... [https://www.illumina.com/techniques/popular-applications/genome-editing.html]
- 96. Whole-genome sequencing reveals rare off-target ... [https://www.nature.com/articles/s41438-021-00549-4]
- 97. Kinetics and Fidelity of the Repair of Cas9-Induced Double ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5993873/]
- 98. Repair of double-strand breaks induced by CRISPR-Cas9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6390938/]
- 99. CMN Weekly (28 November 2025) - Your Weekly CRISPR ... [https://crisprmedicinenews.com/news/cmn-weekly-28-november-2025-your-weekly-crispr-medicine-news/]
- 100. ZFN, TALEN and CRISPR/Cas-based methods for genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3694601/]
- 101. Precision gene editing: The power of CRISPR-Cas in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590234/]
- 102. Mechanism and Applications of CRISPR/Cas-9-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8388126/]
- 103. TALENs—an indispensable tool in the era of CRISPR [https://www.sciencedirect.com/science/article/pii/S1687157X23006571]
- 104. Recent advances in the use of ZFN-mediated gene editing ... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/442/recent-advances-in-the-use-of-zfnmediated-gene-editing-for-human-gene-therapy]
- 105. CRISPR Guide [https://www.addgene.org/guides/crispr/]
- 106. Recent advances in therapeutic gene-editing technologies [https://www.sciencedirect.com/science/article/pii/S152500162500200X]
- 107. Expanding the frontiers of genome engineering [https://www.sciencedirect.com/science/article/abs/pii/S0734975024001757]
- 108. Integration of therapeutic cargo into the human genome ... [https://www.nature.com/articles/s41467-025-57416-2]
- 109. CRISPR RNA-guided integrases for high-efficiency ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10583764/]
- 110. First-in-Human Trial Suggests CRISPR-Cas9 Therapy ... [https://www.acc.org/Latest-in-Cardiology/Articles/2025/11/03/16/19/sat-956am-crispr-aha-2025]
- 111. CMN Weekly (26 September 2025) [https://crisprmedicinenews.com/news/cmn-weekly-26-september-2025-your-weekly-crispr-medicine-news/]
- 112. CRISPR/Cas9 Landscape: Current State and Future ... [https://www.mdpi.com/1422-0067/24/22/16077]
- 113. CRISPR/Cas9 therapeutics: progress and prospects [https://www.nature.com/articles/s41392-023-01309-7]
- 114. Precise measurement of CRISPR genome editing ... [https://www.sciencedirect.com/science/article/pii/S2329050125000440]