CRISPR Gene Editing: Principles, Applications, and Optimization for Drug Development
This article provides a comprehensive guide to CRISPR gene-editing technology, tailored for researchers and drug development professionals.
CRISPR Gene Editing: Principles, Applications, and Optimization for Drug Development
Abstract
This article provides a comprehensive guide to CRISPR gene-editing technology, tailored for researchers and drug development professionals. It covers the foundational principles of CRISPR-Cas systems, from its origins as a bacterial immune mechanism to its function as a programmable DNA-editing tool. The content explores advanced methodological applications in functional genomics and therapy development, details practical strategies for troubleshooting common challenges like off-target effects and editing efficiency, and offers a comparative analysis with traditional gene-editing platforms. The goal is to equip scientists with both the theoretical understanding and practical knowledge needed to effectively implement CRISPR in research and translational medicine.
The CRISPR Revolution: From Bacterial Immunity to a Programmable Genome Editor
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and their CRISPR-associated (Cas) proteins function as a sophisticated adaptive immune system in prokaryotes, enabling bacteria and archaea to defend themselves against invasive genetic elements such as viruses and plasmids [1]. This defense system provides a genetic record of past infections, allowing the host to recognize and eliminate recurring threats with remarkable sequence specificity [2]. The discovery that single-celled organisms possess an adaptive, heritable immune system has fundamentally transformed our understanding of microbe-virus interactions and was later repurposed to revolutionize genetic engineering [1].
The CRISPR-Cas system is characterized by specific DNA loci consisting of short, repetitive sequences interspersed with unique "spacer" sequences derived from previous invaders [1]. These loci are accompanied by Cas genes that encode the protein machinery for system function. Comparative genomic analyses reveal that approximately 40% of sequenced bacteria and over 80% of archaea possess at least one CRISPR-Cas system [1], highlighting its widespread importance in prokaryotic biology.
Molecular Mechanism of CRISPR Adaptive Immunity
The adaptive immune function of CRISPR-Cas systems operates through three distinct, sequential stages: adaptation, expression, and interference. These stages work in concert to provide a dynamic defense mechanism that can adapt to new threats and mount targeted responses against recurrent infections.
Stage 1: Adaptation - Capturing Genetic Memories
The adaptation phase, also called spacer acquisition, represents the immunological memory formation stage where the system captures molecular signatures of invading genetic elements. During this stage, specialized Cas proteins, primarily the Cas1-Cas2 integrase complex, recognize and process foreign DNA from invading viruses or plasmids into short fragments called protospacers [1]. This complex then catalyzes the integration of these protospacers as new spacers into the CRISPR array within the host genome [3] [1]. The CRISPR array thereby accumulates a chronological record of infection events, serving as a genetic memory bank.
Recent research has elucidated that bacteria preferentially acquire spacers from viruses in a dormant state (lysogeny) rather than during active replication [2]. During this "viral nap," bacteria have increased opportunity to integrate viral DNA fragments, effectively "vaccinating" themselves against future infections [2]. This spacer acquisition mechanism represents a sophisticated form of molecular immunity, where the host organism adapts to its pathogenic environment at the genetic level.
Stage 2: Expression - Generating Targeting Molecules
In the expression stage, the CRISPR locus undergoes transcription, producing a long precursor CRISPR RNA (pre-crRNA) [1]. This precursor is then processed by Cas proteins into short, mature crRNAs, each containing a single spacer sequence that serves as a guide for target recognition [4] [1]. In certain systems, such as Type II, a trans-activating crRNA (tracrRNA) facilitates crRNA maturation [3]. Each mature crRNA assembles with one or more Cas proteins to form an effector complex, programming it for specific target recognition in the subsequent interference stage.
Stage 3: Interference - Neutralizing Invaders
The final interference stage represents the execution of immune function, where crRNA-guided effector complexes patrol the cell seeking complementary nucleic acid sequences from invading genetic elements [1]. Upon recognizing a target sequence matching its spacer, the effector complex initiates cleavage, leading to degradation of the invading DNA or RNA [4]. Most DNA-targeting systems require recognition of a short protospacer adjacent motif (PAM) flanking the target sequence, which helps distinguish self from non-self DNA and prevents autoimmunity [1]. This destruction neutralizes the threat, completing the adaptive immune response.
Classification of CRISPR-Cas Systems
CRISPR-Cas systems exhibit considerable diversity, leading to their formal classification into types and subtypes based on their genetic architecture, Cas protein composition, and mechanistic features. The following table summarizes the current classification framework based on the most recent comprehensive analysis.
Table 1: Updated Classification of CRISPR-Cas Systems (2025)
| Class | Type | Signature Protein | Target Molecule | Key Features |
|---|---|---|---|---|
| Class 1 (Multi-subunit effector complexes) | I | Cas3 | DNA | Multi-protein cascade complex with separate nuclease [5] [1] |
| III | Cas10 | DNA/RNA | Involves cyclic oligoadenylate (cOA) signaling; targets both nucleic acids [5] | |
| IV | CasDinG (Csf1) | DNA | Variable adaptation modules; some variants cleave DNA [5] | |
| VII | Cas14 | RNA | Metall-β-lactamase (β-CASP) effector nuclease; targets RNA [5] | |
| Class 2 (Single-protein effectors) | II | Cas9 | DNA | Uses tracrRNA; requires NGG PAM; most widely used in biotechnology [3] [4] |
| V | Cas12 | DNA | Single RuvC-like nuclease domain; targets DNA [4] | |
| VI | Cas13 | RNA | Targets RNA rather than DNA; used in diagnostic applications [4] |
The classification continues to expand with ongoing discovery, with the most recent update identifying 2 classes, 7 types, and 46 subtypes, compared to 6 types and 33 subtypes in the previous classification five years ago [5]. This diversity reflects the evolutionary arms race between prokaryotes and their viral predators across different ecological niches.
Experimental Approaches for Studying CRISPR Function
Research into CRISPR-Cas systems employs sophisticated experimental approaches to elucidate molecular mechanisms and explore system functionalities. The following methodology represents a contemporary approach for investigating spacer acquisition dynamics.
Protocol: Investigating Spacer Acquisition from Lysogenic Phages
Background: This protocol is adapted from recent research published in Cell Host & Microbe that examined how bacteria preferentially acquire spacers from dormant viruses [2]. The methodology enables precise analysis of CRISPR adaptation dynamics under controlled infection conditions.
Materials and Reagents:
- Bacterial strain: Streptococcus pyogenes or other CRISPR-containing species
- Phage variants: Wild-type (capable of lysogeny) and genetically engineered mutants locked in lytic cycle
- Growth media: Appropriate sterile liquid and solid media for host strain
- Selection agents: Antibiotics for maintaining selective pressure if needed
- Molecular biology reagents: PCR components, primers, DNA sequencing reagents
- CRISPR array analysis tools: Sequencing primers targeting CRISPR locus flanks
Procedure:
- Culture preparation: Grow separate bacterial cultures to mid-logarithmic phase in appropriate liquid media.
- Phage infection: Infect experimental groups with wild-type phages (capable of lysogeny) and control groups with engineered phages (locked in lytic cycle) at equivalent multiplicity of infection (MOI).
- Recovery and selection: Allow infection to proceed for predetermined duration, then plate surviving cells on solid media to obtain isolated colonies.
- Genetic analysis: a. Ispace genomic DNA from multiple individual colonies for each condition. b. Amplify CRISPR loci using PCR with primers flanking the array. c. Sequence amplified products using Sanger or next-generation sequencing.
- Spacer identification: Bioinformatically identify newly acquired spacers by comparing to uninfected control sequences.
- Spacer source mapping: Align acquired spacer sequences to both phage and host genomes to determine origins.
- Functional validation: Challenge spacer-carrying bacteria with the same phages to evaluate immune protection.
Key considerations: Maintain appropriate controls throughout, including uninfected bacteria and isogenic bacterial strains differing only in CRISPR status. Standardize culture conditions and infection parameters across experiments to ensure reproducibility.
Signaling Pathways in Type III Systems
Type III CRISPR-Cas systems employ sophisticated signaling pathways that extend beyond direct target cleavage. These systems can initiate a generalized antiviral response through secondary messenger signaling.
Diagram 1: Type III CRISPR-Cas Antiviral Response
Some recently identified type III subtypes (III-G and III-H) show reductive evolution, where the polymerase/cyclase domain of Cas10 is inactivated, resulting in loss of cOA signaling capability while retaining DNA targeting function [5]. This reflects the dynamic evolutionary landscape of CRISPR-Cas systems.
Essential Research Reagents and Tools
The following table catalogs key reagents and materials essential for conducting experimental research on native CRISPR-Cas systems, particularly for studying their adaptive immune functions.
Table 2: Essential Research Reagents for Studying Native CRISPR-Cas Systems
| Reagent/Material | Function in Research | Specific Examples/Applications |
|---|---|---|
| Cas1-Cas2 complex | Study of spacer acquisition mechanism | In vitro assays for protospacer integration [1] |
| Bacteriophage libraries | Sources of protospacers for adaptation studies | Investigating spacer acquisition preferences [2] |
| crRNA/tracrRNA molecules | RNA components for effector complex formation | Studying interference stage mechanisms [3] |
| Type III CRISPR systems | Research on cOA signaling pathways | Analyzing secondary messenger antiviral responses [5] |
| High-throughput screening platforms | Characterization of CAST system variants | Measuring activity and specificity of CRISPR-associated transposons [6] |
| Cas protein variants | Engineering enhanced specificity and activity | V-K CAST mutagenesis for improved genome editing [6] |
| Protospacer adjacent motif (PAM) libraries | Understanding target recognition constraints | Determining sequence requirements for interference [1] |
The CRISPR-Cas system represents a remarkable example of sophisticated adaptive immunity in prokaryotic organisms. Its three-stage mechanism—adaptation, expression, and interference—provides bacteria and archaea with a heritable, sequence-specific defense system against mobile genetic elements. The substantial diversity of CRISPR-Cas systems, now classified into 2 classes, 7 types, and 46 subtypes, reflects continuous evolutionary innovation in host-pathogen conflicts. Ongoing research continues to reveal new dimensions of these systems, from molecular details of spacer acquisition to signaling-mediated antiviral responses. This foundational understanding of CRISPR biology not only advances our knowledge of microbial immunity but also provides the essential framework for developing transformative biotechnological applications.
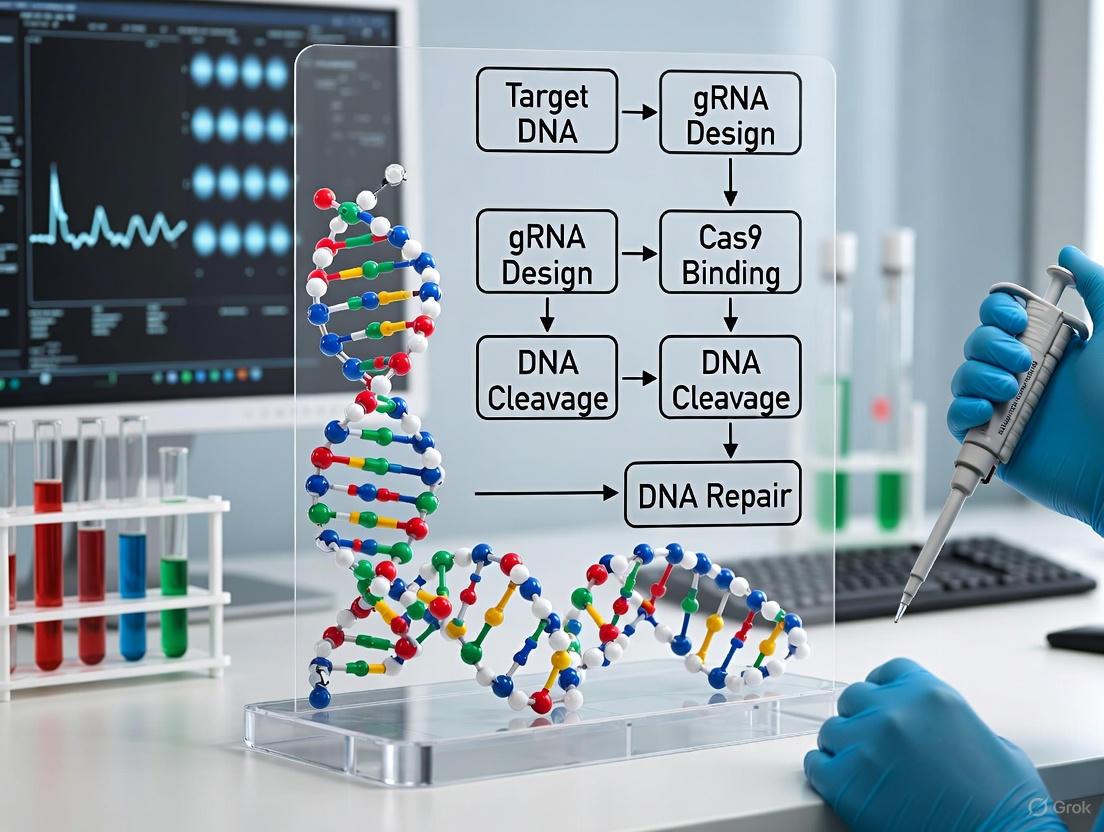
The CRISPR-associated (Cas) protein and guide RNA (gRNA) represent a revolutionary two-component system for precise genome editing. Derived from an adaptive immune system in bacteria, this duo functions as a programmable molecular machine that can be directed to specific locations in a genome to introduce double-stranded breaks in DNA [7] [8]. The core innovation lies in the separation of functions: the Cas protein serves as a nonspecific endonuclease engine, while the gRNA provides the targeting specificity through Watson-Crick base pairing [9] [8]. This modularity has enabled researchers to rapidly adopt and adapt the technology across diverse biological systems, from basic research to therapeutic development. The most extensively characterized system originates from Streptococcus pyogenes (SpCas9), which has become the foundational tool for most CRISPR applications [7] [9].
Structural Architecture of the Cas-gRNA Complex
The Cas9 protein exhibits a bilobed architecture composed of two primary lobes: a recognition lobe (REC) and a nuclease lobe (NUC) [7]. These lobes form a positively charged groove at their interface that accommodates the negatively charged sgRNA:DNA heteroduplex during target recognition and cleavage [7].
Table 1: Domain Architecture of S. pyogenes Cas9
| Lobe | Domain/Region | Residue Range | Primary Function |
|---|---|---|---|
| Recognition (REC) | Bridge Helix | 60-93 | Structural element connecting REC domains |
| REC1 | 94-179, 308-713 | Major interaction site for sgRNA and DNA | |
| REC2 | 180-307 | Non-essential α-helical bundle (can be partially deleted) | |
| Nuclease (NUC) | RuvC Domain | 1-59, 718-769, 909-1098 | Cleaves non-target DNA strand (split into I, II, III motifs) |
| HNH Domain | 775-908 | Cleaves target DNA strand complementary to gRNA | |
| PAM-Interacting (PI) | 1099-1368 | Recognizes protospacer adjacent motif (PAM) sequence |
The REC lobe is predominantly responsible for binding both the sgRNA and the target DNA, while the NUC lobe contains the catalytic centers for DNA cleavage and the domain responsible for recognizing the protospacer adjacent motif (PAM) [7]. The HNH domain, which cleaves the DNA strand complementary to the gRNA, exhibits significant conformational flexibility, which may be crucial for its catalytic activity [7].
Figure 1: Structural organization of the Cas9 protein and its core components. The protein comprises two primary lobes that form a groove for nucleic acid binding.
Composition and Structure of the Guide RNA
The guide RNA exists in multiple formats, with the single guide RNA (sgRNA) being the most common for research applications. The sgRNA is a chimeric RNA molecule created by fusing two natural RNA components: the CRISPR RNA (crRNA) and the trans-activating crRNA (tracrRNA) [9] [10].
Table 2: Components of the Single Guide RNA (sgRNA)
| Component | Length | Function | Key Characteristics |
|---|---|---|---|
| crRNA segment | 17-23 nt | Target recognition via complementarity | 5' end; defines targeting specificity |
| Linker loop | Variable | Connects crRNA and tracrRNA | Synthetic tetra-loop in engineered systems |
| tracrRNA scaffold | ~65 nt | Cas9 protein binding | Forms core duplex RNA structure |
The crRNA portion contains the customizable 17-20 nucleotide "spacer" sequence that determines DNA targeting specificity through complementarity, while the tracrRNA portion serves as a binding scaffold for the Cas9 protein [9] [8]. This synthetic fusion simplifies the experimental system from three components (Cas9, crRNA, tracrRNA) to two components (Cas9, sgRNA), significantly enhancing its utility for genetic engineering [10].
Functional Mechanism of DNA Targeting and Cleavage
Target Recognition and Activation Cycle
The Cas9-sgRNA complex employs a sophisticated multi-step mechanism to locate and cleave its DNA target. The process begins with PAM recognition, followed by DNA melting, seed sequence annealing, and culminating in double-strand break formation.
Figure 2: Functional workflow of Cas9-sgRNA complex in DNA target recognition and cleavage.
The PAM sequence, typically 5'-NGG-3' for SpCas9, serves as an essential binding signal that must be present immediately adjacent to the target sequence [8]. This requirement prevents the CRISPR system from targeting the bacterial genome's own CRISPR arrays. Once the PAM is recognized, the Cas9 protein unwinds the DNA duplex, allowing the seed region (positions 8-10 at the 3' end of the gRNA targeting sequence) to initiate pairing with the target DNA [8]. If sufficient complementarity exists, complete heteroduplex formation occurs, triggering conformational changes that activate the nuclease domains [7].
DNA Cleavage Mechanism
Upon successful target recognition, the Cas9 enzyme introduces a double-strand break (DSB) in the target DNA through the coordinated activity of its two nuclease domains:
- The RuvC domain cleaves the non-complementary DNA strand
- The HNH domain cleaves the complementary DNA strand that is base-paired with the gRNA [7]
This cleavage occurs approximately 3-4 nucleotides upstream of the PAM sequence, resulting in a blunt-ended double-strand break [8]. The cell then repairs this break through either the error-prone non-homologous end joining (NHEJ) pathway, often resulting in insertions or deletions (indels) that disrupt gene function, or the precise homology-directed repair (HDR) pathway when a repair template is provided [8].
Design Principles for Optimal gRNA Performance
Sequence-Based Design Considerations
Effective gRNA design is critical for successful genome editing experiments. Key sequence characteristics significantly impact both on-target efficiency and off-target specificity.
Table 3: Key Parameters for Effective gRNA Design
| Parameter | Optimal Range/Value | Impact on Activity | Rationale |
|---|---|---|---|
| GC Content | 40-80% (ideal: 40-60%) | High GC increases stability but may reduce activity | Overly stable gRNA:DNA duplexes impair cleavage [10] |
| Seed Region GC | Moderate (avoid extremes) | Critical for target recognition | Mismatches in seed region (positions 8-10) abolish cleavage [8] |
| Position 20 | Guanosine (G) preferred | Enhanced cleavage efficiency | Adjacent to PAM; G improves initiation [11] |
| Repetitive Bases | Avoid GGGG, UUU stretches | Prevents synthesis issues and structural problems | G-quadruplex formation; UUU acts as Pol III terminator [10] |
Structural Considerations and gRNA Accessibility
Beyond sequence composition, the secondary structure of the gRNA itself plays a crucial role in determining functionality. Structural accessibility, particularly at the 3' end of the guide sequence (positions 18-20), is a hallmark of highly active sgRNAs [10]. Nucleotides in this seed region must remain unpaired and accessible for efficient target recognition. Inactive gRNAs often form internal hairpin structures where the 3' end of the guide sequence pairs with nucleotides in the tracrRNA scaffold (positions 51-53), creating an extended stem-loop that prevents target DNA binding [11] [10].
Thermodynamic analyses reveal that non-functional guide sequences have significantly higher self-folding free energy (ΔG = -3.1) compared to functional ones (ΔG = -1.9), indicating greater propensity to form internal secondary structures that impair function [10]. Similarly, the stability of the gRNA:DNA heteroduplex affects activity, with non-functional guides forming more stable duplexes (ΔG = -17.2) than functional ones (ΔG = -15.7) [10].
Experimental Protocols for CRISPR-Cas9 Workflows
Protocol 1: Mammalian Cell Genome Editing Using Plasmid-Based Delivery
This standard protocol enables robust gene knockout in human cell lines through NHEJ-mediated repair.
Materials Required:
- Cas9 expression plasmid (e.g., pX458, Addgene #48138)
- sgRNA expression vector or oligos for cloning
- Target cell line (e.g., HEK293T, HeLa)
- Transfection reagent (e.g., Lipofectamine 3000)
- Antibiotics for selection (e.g., puromycin)
- Lysis buffer for genomic DNA extraction
- PCR reagents for amplification of target locus
- Gel electrophoresis equipment or T7 Endonuclease I for mutation detection
- Sequencing primers for validation
Methodology:
- sgRNA Design: Select 20-nucleotide target sequence adjacent to 5'-NGG-3' PAM using design tools (CHOPCHOP, Synthego). Verify specificity via BLAST against relevant genome.
- Cloning: Anneal and phosphorylate oligos encoding sgRNA target, ligate into BbsI-digested Cas9 plasmid. Transform competent E. coli, screen colonies by PCR/sequencing.
- Cell Transfection: Plate 2×10^5 cells/well in 6-well plate 24h before transfection. Transfect with 2μg plasmid DNA using appropriate transfection reagent per manufacturer's protocol.
- Selection/Enrichment: Apply puromycin (1-5μg/mL) 48h post-transfection for 72h (for pX458) or sort GFP-positive cells by FACS.
- Analysis: Harvest cells 5-7 days post-transfection. Extract genomic DNA, amplify target locus by PCR (300-500bp product). Detect indels via T7E1 assay (5-10% agarose gel) or Sanger sequencing with TIDE decomposition analysis.
- Validation: Clone PCR products into TA vector, transform bacteria, sequence 20+ colonies to determine precise editing efficiency and mutation spectrum.
Troubleshooting:
- Low efficiency: Optimize sgRNA design, verify Cas9/sgRNA expression, test alternative transfection methods
- High toxicity: Reduce DNA amount, use inducible Cas9 system, employ Cas9 nickase pairs
- Off-target effects: Use truncated sgRNAs, high-fidelity Cas9 variants, validate potential off-target sites
Protocol 2: In Vitro Cleavage Assay for gRNA Validation
This biochemical approach directly tests sgRNA activity before cell-based experiments, saving time and resources.
Materials Required:
- Purified Cas9 protein (commercial or recombinant)
- In vitro transcribed or synthetic sgRNA
- Target DNA substrate (PCR-amplified genomic region or plasmid)
- Nuclease-free buffer components
- Agarose gel electrophoresis system
- DNA staining dye (e.g., SYBR Safe)
Methodology:
- Ribonucleoprotein (RNP) Complex Formation: Incubate 100nM Cas9 with 120nM sgRNA in reaction buffer (20mM HEPES pH 7.5, 150mM KCl, 10mM MgCl₂, 5% glycerol) for 10min at 37°C.
- Cleavage Reaction: Add 10nM target DNA substrate to RNP complex, incubate at 37°C for 1h in thermal cycler.
- Reaction Termination: Add Proteinase K (0.5mg/mL) with 0.1% SDS, incubate 15min at 56°C.
- Analysis: Resolve cleavage products on 2% agarose gel, stain with DNA dye, visualize under UV light. Quantify band intensity using ImageJ software.
- Kinetic Analysis (Optional): Perform time-course experiments (0-120min) with aliquots taken at intervals to determine cleavage rate.
Expected Results: Successful cleavage produces two DNA fragments from a single substrate. Calculate cleavage efficiency as percentage of cleaved DNA relative to total DNA. Compare EC₅₀ values (Cas9 concentration for half-maximal cleavage) between different sgRNAs [11].
Advanced Engineering and Specificity Enhancements
Engineered Cas9 Variants for Improved Specificity
Wild-type SpCas9 can tolerate mismatches between the gRNA and target DNA, leading to potential off-target effects. Several engineered high-fidelity Cas9 variants have been developed to address this limitation.
Table 4: Engineered Cas9 Variants with Enhanced Specificity
| Variant | Mutations | Mechanism of Action | Applications |
|---|---|---|---|
| eSpCas9(1.1) | K848A, K1003A, R1060A | Weakenes non-target strand interactions | Reduced off-target cleavage while maintaining on-target activity |
| SpCas9-HF1 | N497A, R661A, Q695A, Q926A | Disrupts interactions with DNA phosphate backbone | High-fidelity editing with minimal off-target effects |
| HypaCas9 | N692A, M694A, Q695A, H698A | Enhances proofreading and mismatch discrimination | Improved specificity while maintaining robust on-target editing |
| evoCas9 | M495V, Y515N, K526E, R661Q | Decreased off-target effects through directed evolution | High-fidelity applications in therapeutic development |
| Sniper-Cas9 | F539S, M763I, K890N | Reduced off-target activity with truncated gRNAs | Compatible with 17-18nt gRNAs for enhanced specificity |
These high-fidelity variants typically achieve reduced off-target activity by introducing mutations that weaken Cas9's interactions with the DNA backbone or enhance its ability to discriminate against mismatched targets [8]. While some variants exhibit reduced on-target activity, they provide significantly improved specificity profiles for applications where off-target editing is a major concern.
Alternative Cas Enzymes and Expanding Targeting Range
Beyond SpCas9, numerous alternative Cas enzymes with distinct properties have been characterized and employed for specialized applications:
- SaCas9: From Staphylococcus aureus, recognizes 5'-NNGRR(N)-3' PAM, smaller size for AAV delivery
- Cas12a (Cpf1): Creates staggered DNA cuts, requires only crRNA (no tracrRNA), processes its own CRISPR array
- Cas13: RNA-targeting enzyme for transcriptome engineering and diagnostics
- Engineered PAM-flexible variants: xCas9 and SpCas9-NG recognize NG PAMs, SpRY recognizes NRN/NYN PAMs [8]
These alternatives expand the targeting range of CRISPR systems and enable new applications beyond DNA editing, including RNA targeting, base editing, and epigenetic modification.
The Scientist's Toolkit: Essential Research Reagents
Table 5: Essential Research Reagents for CRISPR-Cas9 Experiments
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Cas9 Expression Systems | pSpCas9(BB)-2A-Puro (PX459), pSpCas9(BB)-2A-GFP (PX458) | Provides Cas9 and sgRNA expression with selection marker | Plasmid format enables stable integration and long-term expression |
| High-Fidelity Cas9 Variants | eSpCas9(1.1), SpCas9-HF1 plasmids | Reduces off-target effects while maintaining on-target activity | Critical for therapeutic applications and sensitive genetic screens |
| sgRNA Synthesis | Synthetic sgRNA, IVT sgRNA kits, sgRNA expression vectors | Delivers targeting component | Synthetic sgRNA offers highest purity and consistency [9] |
| Delivery Vehicles | Lipofectamine 3000, Lentiviral particles, Electroporation systems | Introduces CRISPR components into cells | Choice depends on cell type: chemical (easy), viral (efficient), electroporation (challenging cells) |
| Detection & Validation | T7 Endonuclease I, Surveyor Mutation Detection Kit, Tracking Indels by Decomposition (TIDE) | Identifies successful genome editing | T7E1 for quick assessment; sequencing for precise characterization |
| Bioinformatics Tools | CHOPCHOP, CRISPRscan, Cas-OFFinder, Synthego Design Tool | Designs sgRNAs and predicts off-target sites | Web-based tools streamline design process and improve success rates [12] |
The Cas protein and guide RNA represent one of the most transformative duos in modern molecular biology. Their modular nature - with Cas providing the catalytic engine and gRNA conferring targeting specificity - has democratized genome engineering across biological disciplines. Understanding the structural basis of their interaction, the mechanistic details of DNA recognition and cleavage, and the principles governing gRNA design is fundamental to harnessing their full potential. As the field advances with engineered variants offering enhanced specificity and novel Cas enzymes expanding the targeting landscape, this core technology continues to evolve, offering increasingly sophisticated tools for basic research and therapeutic development. Proper implementation requires careful attention to gRNA design, appropriate controls for specificity validation, and selection of optimal Cas variants for each application.
The CRISPR-Cas9 system has revolutionized genetic research and therapeutic development by providing unprecedented control over genomic manipulation. At the core of this technology lies a fundamental process: the creation of programmed DNA double-strand breaks (DSBs). These breaks serve as the initiating event that enables virtually all subsequent genome engineering outcomes, from simple gene knockouts to precise nucleotide substitutions. The molecular scissors analogy aptly describes the Cas9 endonuclease, which precisely targets and cleaves DNA at user-defined locations [13] [8]. For researchers and drug development professionals, a thorough understanding of DSB creation mechanisms is essential for designing effective experiments and anticipating potential cellular responses, including unintended recombination events and repair pathway choices that impact experimental outcomes and therapeutic safety [13].
This technical guide examines the molecular mechanisms underlying CRISPR-induced DSBs, the resulting cellular repair pathways, and practical experimental considerations for controlling editing outcomes. We present this information within the context of basic CRISPR principles, providing a foundation for researchers to design more precise and efficient genome editing experiments.
Molecular Mechanism of CRISPR-Cas9 Induced DNA Cleavage
The Core CRISPR-Cas9 Complex
The CRISPR-Cas9 system consists of two fundamental components: a Cas9 endonuclease and a guide RNA (gRNA) [8]. The gRNA is a short synthetic RNA composed of a scaffold sequence necessary for Cas-binding and a user-defined ∼20-nucleotide spacer that defines the genomic target to be modified [8]. The ability to change the genomic target of the Cas enzyme by simply changing the target sequence present in the gRNA makes CRISPR highly scalable compared to previous genome engineering technologies [8].
The Cas9 enzyme originates from microbial adaptive immune systems and functions as a programmable DNA endonuclease. The most commonly used variant is SpCas9 from Streptococcus pyogenes [8]. Cas9 undergoes significant conformational changes during its operation: gRNA binding induces a shift into an active, DNA-binding configuration, with the spacer region of the gRNA left free to interact with target DNA [8].
Target Recognition and Cleavage Dynamics
Cas9 recognizes target DNA sequences through a two-step verification process. First, the enzyme identifies a specific protospacer adjacent motif (PAM) sequence adjacent to the target site [8]. For SpCas9, the PAM sequence is 5'-NGG-3', where "N" represents any nucleotide [8]. This PAM requirement is a critical consideration in gRNA design, as an NGG sequence must be positioned correctly near the target site.
Once Cas9 binds a putative DNA target, the seed sequence (8–10 bases at the 3' end of the gRNA targeting sequence) begins to anneal to the target DNA [8]. If the seed and target DNA sequences match, the gRNA continues to anneal to the target DNA in a 3' to 5' direction [8]. The location of any potential mismatches significantly impacts cleavage efficiency—mismatches between the target sequence in the 3' seed sequence inhibit target cleavage, while mismatches toward the 5' end distal to the PAM often permit target cleavage [8].
Upon successful target binding, Cas9 undergoes a second conformational change where its nuclease domains, RuvC and HNH, cleave opposite strands of the target DNA [8]. This results in a double-strand break within the target DNA, located ∼3–4 nucleotides upstream of the PAM sequence [8]. The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the non-complementary strand, generating a blunt-ended DSB [8].
Figure 1: CRISPR-Cas9 Target Recognition and Cleavage Mechanism. The process initiates with PAM recognition, followed by seed sequence annealing, full gRNA hybridization, Cas9 conformational change, and culminating in RuvC and HNH nuclease domain activation to create a blunt-ended double-strand break.
Comparison of Cas Nucleases and Their Cleavage Properties
While Cas9 remains the most widely used enzyme, several other Cas nucleases offer distinct properties, particularly regarding their PAM requirements and cleavage patterns. Cas12a (Cpf1), for example, recognizes T-rich PAM sequences located at the 5' end of the target and creates staggered ends with 5' overhangs, unlike the blunt ends produced by Cas9 [14]. Additionally, Cas12a requires only a CRISPR RNA (crRNA) for activity, while Cas9 requires both crRNA and trans-activating crRNA (tracrRNA) [14].
Table 1: Comparison of Key Cas Nuclease Properties for DSB Creation
| Property | Cas9 | Cas12a (Cpf1) | Cas9 Nickase (D10A) |
|---|---|---|---|
| PAM Sequence | 3'-NGG-5' | 5'-TTN-3' | 3'-NGG-5' |
| Cleavage Pattern | Blunt ends | Staggered ends (5' overhangs) | Single-strand nick |
| gRNA Requirement | crRNA + tracrRNA | crRNA only | crRNA + tracrRNA |
| Active Nuclease Domains | RuvC & HNH | Single RuvC-like | HNH only (RuvC inactivated) |
| DSB Formation | Single enzyme | Single enzyme | Requires two nickases in close proximity |
Cellular Repair Pathways for Double-Strand Breaks
Non-Homologous End Joining (NHEJ)
The efficient but error-prone non-homologous end joining pathway represents the primary repair mechanism for DSBs in most mammalian cells [8]. This pathway directly ligates the broken DNA ends without requiring a homologous template [8]. While efficient in repairing DSBs, NHEJ frequently introduces small nucleotide insertions or deletions (indels) at the DSB site [8]. A population of cells expressing Cas9 and a gRNA will therefore result in a diverse array of mutations [8].
In most CRISPR applications, NHEJ gives rise to small indels in the target DNA that result in amino acid deletions, insertions, or frameshift mutations leading to premature stop codons within the open reading frame (ORF) of the targeted gene [8]. The ideal result is a loss-of-function mutation within the targeted gene, making NHEJ the preferred pathway for gene knockout experiments [8].
Homology-Directed Repair (HDR)
The less efficient but high-fidelity homology-directed repair pathway relies on copying DNA from a matching template to accurately repair or fill in the missing sequence [8]. HDR is more commonly used for precision edits where specific nucleotide changes are required [8]. This pathway is active primarily during the S and G2 phases of the cell cycle when sister chromatids are available as templates [13].
For refined and precise genome editing purposes, HDR is harnessed to copy a specific DNA template (single-stranded or double-stranded) into the target site [13]. The HDR pathway requires a donor template with homologous sequences flanking the target site, which serves as a repair template [8]. While HDR offers greater precision, its lower efficiency compared to NHEJ presents a significant challenge for researchers attempting to introduce specific edits.
DNA Resection: The Critical Decision Point
DNA resection represents a universal process in genome maintenance where one strand of DNA is degraded, leaving the other strand intact [15]. This sometimes highly processive degradation is critical for many forms of DNA damage repair, replication-coupled repair, and meiotic recombination [15]. The resection process must be tightly regulated to prevent genome instability and promote faithful and accurate repair [15].
Resection at DSBs is initiated by the combined action of the Mre11-Rad50-Nbs1 (MRN) complex with its cofactor CtIP and represents one of the first control steps directing repair toward HDR [15]. Following short-range DNA resection, the MRN complex recruits a large molecular machine called the resectosome, a complex comprising a helicase (BLM or WRN), a nuclease (EXO1 or DNA2), and the ssDNA-binding protein replication protein A (RPA) [15]. Whereas short-range DNA resection ranges from tens to hundreds of nucleotides, long-range DNA resection can resect up to 3.5 kb from the DNA break in humans [15].
Figure 2: DNA Double-Strand Break Repair Pathways. Following DSB creation, cells initiate repair through competing pathways. Limited resection directs repair toward error-prone NHEJ, while extensive resection enables high-fidelity HDR using the resectosome complex.
Advanced CRISPR Systems for Controlled DSB Creation
Engineered Cas Variants for Enhanced Specificity
Wild-type Cas9 produces DSBs at both intended and unintended (off-target) sites, posing significant challenges for therapeutic applications. To address this limitation, researchers have developed several engineered Cas9 variants with enhanced specificity:
- High-Fidelity Cas9 (hfCas9): eSpCas9(1.1) weakens interactions between the HNH/RuvC groove and the non-target DNA strand; SpCas9-HF1 disrupts Cas9's interactions with the DNA phosphate backbone; HypaCas9 increases Cas9 proofreading and discrimination [8].
- PAM-Flexible Cas9: xCas9 and SpCas9-NG recognize NG PAMs; SpG recognizes NGN PAMs; SpRY recognizes NRN and NYN PAMs, greatly expanding targeting range [8].
- Cas9 Nickase (Cas9n): A D10A mutant of SpCas9 with one active nuclease domain that generates single-strand breaks rather than DSBs [8]. Using paired nickases (dual nicking) increases specificity as two off-target nicks are unlikely to occur close enough to generate a DSB [8].
DSB-Independent CRISPR Systems
Recent advances have developed CRISPR systems that modify DNA without creating DSBs, addressing safety concerns associated with DSB repair:
- Base Editing: Utilizes a catalytically impaired Cas protein (dCas9 or nickase) fused to a DNA deaminase enzyme to directly convert one base pair to another without DSBs [16]. Cytidine Base Editors (CBEs) convert C•G to T•A, while Adenine Base Editors (ABEs) convert A•T to G•C [16].
- Prime Editing: Employs a Cas9 nickase fused to reverse transcriptase and a specialized prime editing guide RNA (pegRNA) to directly write new genetic information into a target DNA site without DSBs [16]. This system can achieve all 12 possible base-to-base conversions, plus small insertions and deletions [16].
- CRISPR Interference (CRISPRi): Uses catalytically dead Cas9 (dCas9) to bind target DNA without cleavage, blocking transcription and reversibly repressing gene expression [17] [16]. When fused to transcriptional repressor domains like KRAB, CRISPRi can achieve potent gene silencing [17].
Table 2: Comparison of CRISPR Editing Platforms and Their DNA Modification Approaches
| Editing Platform | DSB Formation | Primary Repair Pathway | Editing Outcomes | Therapeutic Applications |
|---|---|---|---|---|
| CRISPR-Cas9 Nuclease | Yes | NHEJ/HDR | Indels, precise edits with donor | Gene knockouts, gene correction |
| Base Editing | No | DNA mismatch repair | Point mutations | Correcting point mutations causing disease |
| Prime Editing | No | DNA repair synthesis | All 12 base changes, small indels | Precise editing without donor templates |
| CRISPRi/a | No | N/A | Gene expression modulation | Gene regulation without DNA sequence change |
Experimental Considerations and Methodologies
Designing and Validating CRISPR Experiments
Successful CRISPR editing requires careful experimental design and validation. When designing gRNAs, researchers should select target sequences with perfect homology to the target DNA and minimal homology elsewhere in the genome to reduce off-target effects [8]. Various online tools are available to help select optimized gRNAs for specific applications [8].
For HDR-based editing, researchers must design donor templates with sufficient homology arms flanking the desired edit. Studies suggest that asymmetric homology arms with longer 5' arms may improve HDR efficiency in some systems. Additionally, timing Cas9 expression with cell cycle phases when HDR is active (S/G2) can improve precise editing efficiency.
Analyzing Editing Outcomes
Validating CRISPR editing is a critical step in any genome engineering experiment. Several methods are available to assess CRISPR editing efficiency:
- Next-Generation Sequencing (NGS): The gold standard for analyzing CRISPR editing outcomes, providing comprehensive view of indels generated with high sensitivity [18]. Best suited for large-scale studies with bioinformatics support.
- Inference of CRISPR Edits (ICE): Uses Sanger sequencing data to determine relative abundance and levels of indels [18]. Highly comparable to NGS (R² = 0.96) but more accessible for smaller labs.
- Tracking of Indels by Decomposition (TIDE): An older decomposition method using Sanger sequencing data to estimate relative abundance of insertions or deletions [18]. Limited primarily to detecting +1 insertions.
- T7 Endonuclease 1 (T7E1) Assay: A non-sequencing based method that detects mismatched DNA heteroduplexes [18]. Least expensive but provides no sequence-level information.
The Scientist's Toolkit: Essential Reagents and Methods
Table 3: Essential Research Reagents and Methods for CRISPR DSB Research
| Reagent/Method | Function | Application Notes |
|---|---|---|
| Cas9 Expression Vector | Expresses Cas9 endonuclease in target cells | Choose between wild-type, nickase, or dead Cas9 depending on application |
| Guide RNA Cloning Vector | Expresses sequence-specific gRNA | Enables multiplexing with multiple gRNAs for complex edits |
| Donor Template | Provides homology for HDR repair | Single-stranded or double-stranded DNA with homology arms |
| Delivery Method | Introduces CRISPR components into cells | Viral (lentiviral, AAV), electroporation, or transfection methods |
| Validation Primers | Amplify target locus for analysis | Design to flank cut site by 100-300bp for optimal resolution |
| Next-Generation Sequencing | Comprehensive analysis of editing outcomes | Provides deepest analysis but requires bioinformatics expertise |
Unintended Consequences and Safety Considerations
Chromosomal Rearrangements and Recombination Events
CRISPR-induced DSBs may trigger unintended genetic consequences beyond small indels. Studies in Drosophila demonstrate that Cas9-mediated editing events frequently result in germline-transmitted exchange of chromosome arms—often without indels [13]. These recombination events occurred in up to 39% of detected CRISPR events, even on chromosomes that normally exhibit no native recombination activity [13].
Such findings highlight an unforeseen risk of using CRISPR-Cas9 for therapeutic intervention, as large-scale chromosomal rearrangements could have significant pathological consequences [13]. The ability of CRISPR-Cas9 to introduce several concurrent DSBs at defined positions has been leveraged to engineer tumor-associated chromosomal translocations resembling those observed in cancers [13].
Mitigating Off-Target Effects
Several strategies have been developed to minimize off-target editing:
- High-Fidelity Cas Variants: eSpCas9(1.1), SpCas9-HF1, and HypaCas9 exhibit reduced off-target activity while maintaining robust on-target editing [8].
- Dual Nickase Approach: Using two Cas9 nickases with offset gRNAs requires both gRNAs to bind in close proximity to generate a DSB, dramatically increasing specificity [8].
- Computational gRNA Design: Careful gRNA selection to avoid sequences with high similarity to other genomic regions reduces off-target potential [8].
- Truncated gRNAs: Using shorter gRNAs (17-18 nt instead of 20 nt) can increase specificity, though with potential reduction in on-target efficiency [8].
- RIGS (Reference-based In silico Genome Screening): In silico methods to predict and avoid gRNAs with potential off-target sites [19].
The creation of programmed double-strand breaks represents the foundational mechanism underlying CRISPR-Cas9 genome editing. Understanding the molecular details of DSB formation, cellular repair pathways, and potential unintended consequences enables researchers to design safer and more effective experiments. As CRISPR technologies evolve toward DSB-free editing systems like base editing and prime editing, the fundamental principles of target recognition and DNA modification remain essential knowledge for researchers and therapeutic developers. The continued refinement of CRISPR systems promises to enhance both the precision and safety of genome engineering for basic research and clinical applications.
The CRISPR-Cas9 system has revolutionized genetic research by providing scientists with unprecedented precision in genome editing. However, the CRISPR-Cas9 enzyme itself functions merely as a pair of "molecular scissors" that creates a specific double-strand break (DSB) in the DNA [20]. The actual genetic modification occurs through the cell's endogenous DNA Damage Repair (DDR) pathways, which are activated to join the two cut ends [20] [21]. These pathways are essential for maintaining genomic integrity across all organisms and represent a collection of intracellular mechanisms that sense DNA damage, alert the cell to its presence, and prompt repair [20]. For researchers aiming to leverage CRISPR technology, understanding how to harness two key competing repair pathways—Non-Homologous End Joining (NHEJ) and Homology-Directed Repair (HDR)—is fundamental to achieving specific editing outcomes, whether for gene knockouts, precise point mutations, or gene knockins [20] [21].
Table 1: Core Characteristics of NHEJ and HDR Pathways
| Feature | Non-Homologous End Joining (NHEJ) | Homology-Directed Repair (HDR) |
|---|---|---|
| Template Requirement | No template required; uses broken ends [20] | Requires homologous donor template (e.g., sister chromatid, ssODN, plasmid) [20] [22] |
| Primary Mechanism | Direct ligation of DNA ends [23] | Uses homologous sequence as a template for accurate repair [22] |
| Fidelity | Error-prone; often results in small insertions or deletions (INDELs) [20] [21] | High-fidelity and precise [22] |
| Cell Cycle Phase | Active throughout all cell cycle phases [23] | Primarily active in S and G2 phases [20] [24] |
| Primary Application in CRISPR | Gene knockouts (disruption of gene function) [20] [21] | Gene knockins, precise point mutations, inserting fluorescent tags [20] [21] [23] |
| Typical Efficiency | High efficiency (fast and predominant pathway) [20] [24] | Low to moderate efficiency (requires competing with NHEJ) [23] [24] |
DNA Repair Pathway Mechanisms
Non-Homologous End Joining (NHEJ): The Quick Fix
NHEJ is a faster, more efficient repair pathway that operates by quickly rejoining the broken DNA ends without the need for a homologous template [20]. This speed comes at the cost of precision, as the process often leads to small insertions or deletions (INDELs) at the repair site [20] [21]. The term "non-homologous" refers to the fact that the two broken ends are indiscriminately ligated back together with minimal reference to the original DNA sequence [20]. The mechanism initiates with the Ku heterodimer (Ku70/Ku80) recognizing and binding to the DSB ends [23]. This Ku-DNA complex then recruits other core NHEJ factors, including the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and the endonuclease Artemis, which may process the DNA ends [23]. Finally, the XRCC4-DNA ligase IV complex catalyzes the ligation of the DNA ends, often resulting in the characteristic INDEL mutations [23]. While these errors are undesirable in many contexts, they are ideal for gene knockout studies where the goal is to disrupt a gene's function by introducing frameshift mutations or premature stop codons [20].
Homology-Directed Repair (HDR): Precision Engineering
In contrast to NHEJ, HDR is a precise DNA repair mechanism that utilizes a homologous DNA sequence as a template to accurately repair the DSB [21] [22]. This template can be a sister chromatid, a donor plasmid, or a single-stranded oligodeoxynucleotide (ssODN) [21] [22]. The HDR process involves several key steps: first, the 5' ends at the break site are resected by nucleases to create 3' single-stranded DNA (ssDNA) overhangs [22]. This ssDNA then invades a homologous donor template—a step facilitated by recombinase enzymes—and uses the homologous regions as a blueprint for repair synthesis [22]. There are several sub-pathways of HDR, including Synthesis-Dependent Strand Annealing (SDSA) and Double-Strand Break Repair (DSBR), which ultimately resolve to complete the repair with high fidelity [22]. Because this pathway allows researchers to introduce specific sequences provided by an exogenous donor template, it is the foundation for precise genetic modifications such as gene knockins, point mutations, and the creation of transgenic models [20] [21] [23].
Diagram 1: Competitive DSB Repair Pathways. Following a CRISPR-induced break, cells can repair the damage via the faster, error-prone NHEJ pathway or the precise, template-dependent HDR pathway. The presence of an exogenous donor template is required for HDR.
Experimental Design for Pathway-Specific Editing
Strategic Selection for Genetic Outcomes
Choosing between NHEJ and HDR is dictated by the ultimate experimental goal. NHEJ is the preferred pathway when the objective is to disrupt a gene's function, creating a knockout [20] [21]. The INDELs introduced by NHEJ frequently cause frameshift mutations that lead to premature stop codons, effectively inactivating the gene [20]. This approach is invaluable for loss-of-function studies. Conversely, HDR is indispensable when precision is required [21]. It enables the introduction of specific nucleotide changes, the insertion of reporter genes like GFP for protein localization studies, or the creation of disease-associated point mutations in model organisms [20] [21] [23]. Achieving HDR requires co-delivering a donor template alongside the CRISPR-Cas9 components. The design of this template is critical: for small edits (1-50 bp), single-stranded oligodeoxynucleotides (ssODNs) with 30-50 base homology arms are standard [22]. For larger insertions, such as fluorescent proteins or selection cassettes, double-stranded DNA (dsDNA) plasmids with longer homology arms (500-1000 bp) are typically used [22]. A key consideration in donor design is to disrupt the protospacer adjacent motif (PAM) or the guide RNA binding site to prevent repeated cleavage of successfully edited alleles by Cas9 [22].
Methodologies to Modulate Repair Pathway Balance
A significant challenge in CRISPR editing is that NHEJ is the dominant and more efficient pathway in most eukaryotic cells, often outcompeting HDR [20] [24]. Consequently, researchers have developed strategies to coax cells toward HDR for more precise edits. One common approach is to inhibit the NHEJ pathway chemically or genetically. This can be achieved by using small molecule inhibitors targeting key NHEJ proteins, such as DNA-PKcs inhibitors, or through siRNA-mediated knockdown of factors like Ku70/80 [23]. An alternative strategy is to synchronize cells in the S and G2 phases of the cell cycle, where HDR is naturally more active [24]. Furthermore, the timing of Cas9 and donor template delivery can be optimized; some studies suggest delivering the donor template before the CRISPR machinery to increase its availability at the time of the DSB [23]. Finally, using Cas9 nickases—mutants that cut only one DNA strand—can also promote HDR. By using a pair of nickases to create a DSB with overhangs or by nicking near the intended edit site in the presence of a donor template, the frequency of HDR can be improved while reducing NHEJ-associated indels [23].
Table 2: Reagent Kits and Solutions for NHEJ and HDR Workflows
| Research Reagent | Function/Description | Example Application |
|---|---|---|
| Cas9 Nuclease | Engineered nuclease that creates DSBs at genomic targets specified by a gRNA [25]. | Essential for initiating both NHEJ and HDR repair pathways [20]. |
| Guide RNA (gRNA) | A synthetic RNA complex that directs Cas9 to a specific genomic locus [25]. | Determines the target site for CRISPR cutting; can be produced via in vitro transcription or chemical synthesis [25]. |
| ssODN Donor Template | A single-stranded DNA oligo containing the desired edit, flanked by homology arms [22]. | Used as a repair template for HDR to introduce small, precise edits (e.g., point mutations) [22]. |
| dsDNA Donor Plasmid | A double-stranded DNA plasmid containing the insertion sequence flanked by long homology arms [22]. | Used as a repair template for HDR to insert large sequences (e.g., fluorescent protein genes) [22]. |
| NHEJ Inhibitors (e.g., DNA-PKcs inhibitors) | Small molecules that chemically inhibit key proteins in the NHEJ pathway [23]. | Used to suppress the error-prone NHEJ pathway and increase the relative efficiency of HDR [23]. |
| Cas9 Nickase | A mutant Cas9 (e.g., D10A) that nicks only one DNA strand instead of creating a DSB [23]. | Using a pair of nickases can reduce off-target effects and promote HDR by creating staggered breaks [23]. |
Advanced Applications and Clinical Translation
The fundamental principles of harnessing NHEJ and HDR are driving the next generation of gene therapies and biomedical research. In clinical trials, both pathways are being exploited for therapeutic benefit. A landmark example is Casgevy, the first FDA-approved CRISPR-based medicine for sickle cell disease and transfusion-dependent beta thalassemia, which leverages CRISPR-mediated editing to disrupt (via NHEJ-like mechanisms) the BCL11A gene to reactivate fetal hemoglobin [26]. Meanwhile, HDR-based strategies are advancing toward the clinic for correcting specific pathogenic mutations. In 2025, a historic milestone was achieved with the development of a personalized in vivo CRISPR therapy for an infant with CPS1 deficiency, which was developed, approved, and delivered in just six months [26]. This case serves as a proof-of-concept for on-demand gene-editing therapies for rare genetic diseases. Furthermore, companies like Intellia Therapeutics are demonstrating the success of in vivo HDR-based therapies for diseases like hereditary transthyretin amyloidosis (hATTR), where a single intravenous infusion of CRISPR-LNPs (lipid nanoparticles) leads to a deep and sustained reduction of the disease-related protein in the liver [26]. Beyond traditional CRISPR knockouts, newer modalities like base editing and prime editing are being developed to achieve precise changes without requiring a DSB, thereby bypassing the inherent competition between NHEJ and HDR and potentially increasing safety and efficiency [27] [28].
Diagram 2: Experimental Workflow for Pathway Selection. A decision tree guiding researchers from experimental goal to the appropriate repair pathway strategy and subsequent validation methods.
The conscious application of the NHEJ and HDR DNA repair pathways is a cornerstone of effective CRISPR-Cas9 genome editing. NHEJ offers a straightforward and efficient method for gene disruption, while HDR provides the precision necessary for sophisticated genetic modeling and therapeutic correction. The ongoing development of strategies to bias the cellular repair machinery toward HDR, such as NHEJ inhibition and cell cycle manipulation, coupled with advances in delivery systems like lipid nanoparticles (LNPs), is continuously enhancing the efficacy and expanding the applications of precision editing [26] [23]. As the field progresses, the integration of these fundamental principles with emerging technologies—including AI-powered design tools like CRISPR-GPT and next-generation base editors—promises to further accelerate the development of life-saving genetic therapies and deepen our understanding of biological systems [29] [28]. For researchers, a firm grasp of these cellular repair pathways remains indispensable for translating the cutting potential of CRISPR into meaningful and predictable genetic outcomes.
The discovery of the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system has revolutionized genetic engineering, offering an unprecedented ability to manipulate nucleic acids with high precision. While the CRISPR-associated protein 9 (Cas9) from Streptococcus pyogenes has been the workhorse of genome editing, it represents only one facet of a diverse molecular ecosystem [30]. This review moves beyond the familiar Cas9 to explore the expanding universe of other Cas effectors—particularly Cas12 and Cas13—that have emerged as powerful tools with distinct functionalities. Framed within the broader principles of CRISPR research, this technical guide examines the unique mechanisms, applications, and experimental protocols for these alternative Cas proteins, providing researchers and drug development professionals with the knowledge to leverage these systems for advanced genetic manipulation.
The classification of CRISPR-Cas systems into two classes based on effector complex architecture provides crucial context for understanding these molecular tools. Class 1 systems (types I, III, and IV) utilize multi-protein effector complexes, while Class 2 systems (types II, V, and VI) employ single effector proteins like Cas9, Cas12, and Cas13 [31] [32]. It is within Class 2 that we find the proteins of focus for this review: Cas12 (type V) for DNA targeting and Cas13 (type VI) for RNA targeting, each offering unique advantages that complement and in some cases surpass the capabilities of Cas9 [33].
Classification and Basic Mechanisms
CRISPR-Cas systems function as adaptive immune systems in prokaryotes, providing sequence-specific protection against foreign genetic elements. This adaptive immunity operates through three distinct stages: adaptation, where short DNA fragments from invaders (protospacers) are integrated into the host CRISPR array; expression, involving transcription of the CRISPR array into precursor CRISPR RNA (pre-crRNA) and its processing into mature crRNAs; and interference, where crRNA-guided Cas effector complexes recognize and cleave complementary nucleic acids [31] [32]. This fundamental mechanism forms the foundation for all CRISPR-based technologies, with variations occurring in the specific components and pathways utilized by different Cas proteins.
Classification of Cas Proteins
Table 1: Classification and Key Characteristics of Major Class 2 CRISPR-Cas Effectors
| Effector Protein | CRISPR Type | Class | Target Nucleic Acid | Protospacer Adjacent Motif (PAM) | Cleavage Pattern | Key Domains |
|---|---|---|---|---|---|---|
| Cas9 | II | 2 | dsDNA | 5'-NGG-3' (SpCas9) | Blunt ends | RuvC, HNH |
| Cas12 | V | 2 | dsDNA/ssDNA | 5'-TTN-3' (FnCas12a) | Staggered ends | RuvC-only |
| Cas13 | VI | 2 | RNA | Protospacer Flanking Site (PFS) | RNA cleavage | HEPN |
The classification of Cas proteins reveals their evolutionary relationships and functional capabilities. Cas12 effectors (type V) differ significantly from Cas9 in both structure and mechanism. Unlike Cas9, which requires two nuclease domains (HNH and RuvC) for DNA cleavage, Cas12 proteins utilize a single RuvC domain to cleave both DNA strands, resulting in staggered ends rather than blunt cuts [34] [33]. This structural simplification presents potential advantages for certain applications. Furthermore, Cas12 recognizes distinct T-rich PAM sequences (e.g., 5'-TTN-3'), significantly expanding the targeting range beyond the G-rich PAMs required by SpCas9 [33].
Cas13 effectors represent a fundamentally different class of CRISPR tools as they exclusively target RNA rather than DNA [32]. Cas13 proteins contain two Higher Eukaryotes and Prokaryotes Nucleotide-binding (HEPN) domains responsible for RNA cleavage activity [33]. A unique feature of Cas13 is its collateral activity—upon recognition and cleavage of its target RNA, the protein becomes a promiscuous RNase that non-specifically degrades surrounding RNA molecules [32] [33]. This activity, while potentially a limitation for therapeutic applications, has been harnessed for highly sensitive diagnostic platforms.
Cas12 Family: DNA-Targeting Effectors
Mechanism of Action
Cas12 effectors operate through a distinct mechanism from Cas9. Upon recognition of a target DNA sequence adjacent to a compatible PAM, Cas12 undergoes conformational changes that position its single RuvC domain for sequential cleavage of both DNA strands [33]. The resulting staggered double-strand breaks typically feature 5-8 nucleotide overhangs, unlike the blunt ends produced by Cas9 [34]. This cleavage pattern can be advantageous for certain genome engineering applications where staggered ends potentially enhance homology-directed repair.
Another remarkable property of Cas12 is its trans-cleavage activity. After recognizing and cleaving its target DNA, Cas12 exhibits nonspecific single-stranded DNA (ssDNA) cleavage activity [33]. This activated state persists beyond the initial target recognition, enabling continuous degradation of surrounding ssDNA molecules. This collateral effect has been creatively harnessed for diagnostic applications, forming the basis for sensitive DNA detection platforms.
Subtypes and Applications
The Cas12 family includes several subtypes with distinct characteristics:
Cas12a (Cpf1): The most extensively characterized Cas12 variant, capable of processing its own crRNA arrays without requiring tracrRNA [33]. This feature enables simpler multiplexing approaches where multiple guide sequences can be expressed from a single transcript.
Cas12b: Significantly smaller than Cas9 and Cas12a, making it potentially more suitable for viral delivery [33]. Early versions required elevated temperatures for optimal activity, but engineered variants now function efficiently at mammalian physiological temperatures.
Cas12i and Cas12j: More recently discovered variants with compact sizes that show promise for therapeutic applications due to their high specificity and minimal off-target effects [33].
Table 2: Comparison of Cas12 Subtypes and Their Research Applications
| Cas12 Subtype | Size (aa) | PAM Requirement | crRNA Processing | Key Applications | Advantages for Research |
|---|---|---|---|---|---|
| Cas12a (Cpf1) | ~1300 | 5'-TTN-3' | Autonomous | Genome editing, multiplexed targeting, DNA detection | Simplified multiplexing, staggered cuts for HDR |
| Cas12b | ~1100 | 5'-TTN-3' | Requires RNase | In vivo editing, viral delivery | Small size for AAV packaging, high specificity |
| Cas12i | ~1000 | 5'-TTN-3' | Autonomous | Therapeutic genome editing | High fidelity, minimal off-target effects |
| Cas12j (CasΦ) | ~700-800 | 5'-TTN-3' | Autonomous | Compact delivery, basic research | Ultra-small size, novel phylogenetic origin |
Cas13 Family: RNA-Targeting Effectors
Mechanism of Action
Cas13 represents a paradigm shift in CRISPR technology as the first well-characterized system to specifically target RNA molecules rather than DNA [32]. Cas13 effectors are guided by a single crRNA to complementary RNA sequences and cleave their targets using two HEPN domains that form the active RNase site [33]. Upon target recognition and cleavage, Cas13 undergoes a conformational change that activates its nonspecific collateral RNase activity, leading to degradation of nearby non-target RNA molecules [32] [33].
The Cas13 family includes multiple subtypes (VI-A through VI-D) with variations in crRNA handling and targeting requirements [32]. Unlike DNA-targeting Cas proteins that require specific PAM sequences, Cas13 recognition depends on protospacer flanking sites (PFS) with slight biases toward certain nucleotides but generally less restriction than DNA-targeting systems [32] [33].
Subtypes and Research Utilities
The Cas13 family has been classified into several subtypes with distinct properties:
Cas13a (C2c2): The first characterized RNA-targeting Cas protein, capable of processing pre-crRNA and exhibiting strong collateral activity in bacterial systems [33].
Cas13b: Often found in conjunction with accessory proteins that may modulate its activity, offering potential regulatory control mechanisms [32].
Cas13d: The most compact variant (~930 amino acids), making it suitable for viral delivery and enabling efficient RNA knockdown in eukaryotic cells without significant collateral effects [33].
The primary research applications of Cas13 leverage its programmable RNA-binding capability. These include: Transcript knockdown as an alternative to RNAi with potentially higher specificity; Live RNA imaging using catalytically inactive dCas13 fused to fluorescent proteins; RNA base editing through fusion with adenosine deaminases (e.g., ADAR2) for precise A-to-I conversions; and Nucleic acid detection utilizing the collateral activity for signal amplification in diagnostic platforms [34] [33].
Comparative Analysis and Selection Guide
Functional Comparison
Understanding the distinct properties of Cas9, Cas12, and Cas13 is crucial for selecting the appropriate tool for specific research goals. The following comparative analysis highlights key functional differences:
Table 3: Functional Comparison of Cas9, Cas12, and Cas13 Effectors
| Parameter | Cas9 | Cas12 | Cas13 |
|---|---|---|---|
| Primary Target | dsDNA | dsDNA/ssDNA | RNA |
| Cleavage Pattern | Blunt ends | Staggered ends | RNA fragments |
| Guide RNA | tracrRNA + crRNA | crRNA only | crRNA only |
| crRNA Processing | Requires RNase III | Autonomous | Autonomous |
| Collateral Activity | No | ssDNA cleavage after activation | RNA cleavage after activation |
| PAM/PFS Requirement | 5'-NGG-3' (SpCas9) | 5'-TTN-3' (Cas12a) | Minimal, preference for 3' nucleotide |
| Multiplexing Capacity | Moderate (with engineering) | High (natural crRNA array) | High (natural crRNA array) |
| Therapeutic Applications | Ex vivo cell therapy, gene disruption | Gene editing, DNA detection | RNA knockdown, RNA editing, diagnostics |
Selection Guidelines for Research Applications
Choosing the appropriate Cas protein depends on the specific research objectives:
For permanent DNA modification: Cas9 remains the standard for many applications, while Cas12 offers advantages for multiplexed editing or when targeting T-rich genomic regions.
For transient transcriptional modulation: Cas13 provides a powerful platform for knocking down RNA without altering the genome, ideal for therapeutic applications where temporary effects are desirable.
For diagnostic applications: Both Cas12 and Cas13 excel in nucleic acid detection due to their collateral activities, with Cas12 used for DNA targets and Cas13 for RNA targets.
For viral delivery: Compact variants like Cas12j and Cas13d offer advantages due to their smaller size, enabling packaging into adeno-associated virus (AAV) vectors with larger cargo capacities.
Experimental Design and Protocols
General Workflow for Cas12 Genome Editing
The experimental pipeline for Cas12-mediated genome editing shares similarities with Cas9 protocols but requires consideration of its unique properties:
Target Selection: Identify target sites with appropriate PAM sequences (typically 5'-TTN-3' for Cas12a). Verify specificity using in silico prediction tools to minimize off-target effects.
Guide RNA Design: Design crRNAs with ~20-24 nucleotide spacer sequences. For multiplexed editing, design crRNA arrays with direct repeats separating individual spacer sequences.
Delivery System Preparation:
- For plasmid-based delivery: Clone crRNA expression cassettes into appropriate vectors. Cas12 expression can be driven by constitutive (e.g., EF1α) or inducible promoters.
- For ribonucleoprotein (RNP) delivery: In vitro transcribe crRNA and purify Cas12 protein. Complex at molar ratios of 1:2 to 1:4 (protein:RNA) for 10-30 minutes before delivery.
Delivery into Target Cells:
- For mammalian cells: Use electroporation for RNP delivery or lipid-based transfection for plasmid DNA.
- For primary cells: Optimized RNP delivery typically yields highest editing efficiency with minimal toxicity.
Validation and Analysis:
- Assess editing efficiency 48-72 hours post-delivery using T7E1 assay, TIDE analysis, or next-generation sequencing.
- Evaluate potential off-target effects at predicted sites using targeted sequencing.
Protocol for Cas13-Mediated RNA Knockdown
Cas13 enables programmable RNA knockdown without genomic alteration. The following protocol outlines key steps for implementing this technology:
Target Selection: Identify accessible regions within target transcripts using RNA accessibility prediction tools or empirical testing. Avoid highly structured regions if possible.
crRNA Design: Design crRNAs with spacer sequences complementary to the target RNA. Include 28-30 nucleotides for optimal activity with most Cas13 orthologs.
Expression System Selection:
- For transient expression: Use plasmid vectors with RNA polymerase III promoters (U6) for crRNA expression and polymerase II promoters for Cas13 expression.
- For stable expression: Integrate Cas13 expression cassette into safe-harbor loci (e.g., AAVS1) and deliver crRNAs via viral vectors.
Delivery Optimization:
- For in vitro applications: Transfect plasmid DNA or in vitro transcribed components using lipid-based reagents.
- For in vivo applications: Utilize AAV vectors (serotypes vary by tissue tropism) for delivery of compact Cas13 variants like Cas13d.
Efficiency Validation:
- Quantify knockdown efficiency 24-96 hours post-delivery using RT-qPCR, RNA-seq, or western blot (if protein reduction is measured).
- Monitor potential collateral effects by examining housekeeping gene expression levels.
Phenotypic Assessment:
- Evaluate functional consequences of knockdown using cell-based assays relevant to the target pathway.
- For therapeutic applications, assess rescue of disease-associated phenotypes.
Diagnostic Assay Development with Cas12 and Cas13
The collateral activities of Cas12 and Cas13 have been harnessed for highly sensitive diagnostic platforms. The general workflow for developing such assays includes:
Sample Preparation: Extract nucleic acids from biological samples. For RNA targets, include a reverse transcription step.
Target Amplification: Implement isothermal amplification (e.g., RPA, LAMP) to increase target abundance. This step enhances detection sensitivity to attomolar or zeptomolar levels.
Cas Detection Reaction:
- Program the Cas protein (Cas12 for DNA, Cas13 for RNA) with target-specific crRNAs.
- Include reporter molecules (e.g., fluorescently labeled ssDNA/RNA oligonucleotides) that will be cleaved upon Cas activation.
- Incubate at optimal temperature (typically 37°C) for 30-90 minutes.
Signal Detection:
- Use fluorimeters for quantitative measurements.
- Alternatively, employ lateral flow strips for visual readouts suitable for point-of-care applications.
Validation: Compare with established detection methods (e.g., PCR) to determine sensitivity and specificity.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Reagents for Cas12 and Cas13 Research
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Expression Plasmids | Cas12a/pCAG, Cas13d/U6-gRNA | Protein and guide RNA expression | Choose promoters based on cell type (U6 for crRNA, CAG/EF1α for Cas) |
| Purified Proteins | Recombinant Cas12a, Cas13d | For RNP delivery | Higher specificity, reduced off-target effects compared to plasmid delivery |
| Synthetic crRNAs | Chemically synthesized spacers | Guide RNA for target recognition | Can include modified nucleotides for enhanced stability |
| Detection Reporters | FQ-reporters (ssDNA/RNA) | For collateral activity detection | Essential for diagnostic applications and activity validation |
| Amplification Reagents | RPA/LAMP kits | Pre-amplification for diagnostics | Enables high sensitivity in detection assays |
| Delivery Tools | Electroporation systems, Lipofectamine | Introducing components into cells | RNP delivery preferred for primary cells |
| Validation Kits | T7E1, TIDE, NGS libraries | Editing efficiency quantification | NGS provides most comprehensive analysis |
Emerging Applications and Future Directions
The unique properties of Cas12 and Cas13 proteins have enabled applications beyond traditional genome editing:
Therapeutic Development: Cas13 shows exceptional promise for treating RNA-based diseases. In a landmark 2025 case, researchers developed a personalized in vivo CRISPR therapy for an infant with CPS1 deficiency using LNP-delivered Cas13, demonstrating the potential for rapid development of treatments for rare genetic disorders [26]. Intellia Therapeutics has advanced Cas13-based therapies for hereditary transthyretin amyloidosis (hATTR) and hereditary angioedema (HAE) into clinical trials, showing sustained reduction of disease-related proteins in patients [26].
Diagnostic Platforms: The collateral activities of Cas12 and Cas13 form the foundation of revolutionary diagnostic tools. The DNA Endonuclease-Targeted CRISPR Trans Reporter (DETECTR) system utilizing Cas12a enables attomolar-level DNA detection, while the Specific High-sensitivity Enzymatic Reporter UnLOCKing (SHERLOCK) platform based on Cas13 achieves zeptomolar sensitivity for RNA targets [35] [33]. These systems have been deployed for detecting pathogens including SARS-CoV-2 with point-of-care applicability [35].
Advanced Research Tools: Engineered catalytically inactive versions of these proteins (dCas12, dCas13) serve as programmable DNA- and RNA-binding platforms for applications including base editing, epigenetic modification, live-cell imaging, and transcript tracking [34]. Fusion proteins combining dCas13 with adenosine deaminases (ADAR) enable precise RNA base editing without permanent genomic changes [34].
The future of CRISPR technology beyond Cas9 will likely see increased specialization of Cas proteins for particular applications, enhanced by engineering efforts to improve specificity, efficiency, and delivery. The integration of artificial intelligence tools like CRISPR-GPT is already accelerating experimental design and optimization, potentially reducing development timelines for new therapies [29]. As our understanding of these diverse molecular machines deepens, their impact on basic research and therapeutic development will continue to expand, offering new avenues for manipulating biological systems with unprecedented precision.
Visual Guide to Cas12 and Cas13 Mechanisms
From Bench to Bedside: CRISPR Workflows and Therapeutic Applications
The CRISPR-Cas9 system has revolutionized genetic research and therapeutic development by providing a precise and programmable method for genome editing. At the heart of this technology lies the guide RNA (gRNA), a short nucleic acid sequence that directs the Cas nuclease to specific genomic targets. The effectiveness of CRISPR experiments hinges on the design of highly functional gRNAs that maximize on-target efficiency while minimizing off-target effects. This technical guide explores the fundamental principles of gRNA design, key parameters for optimization, bioinformatics tools for selection, and experimental methodologies for validation. By synthesizing current research and emerging trends, including the application of artificial intelligence in editor design, this review provides researchers with a comprehensive framework for developing effective gRNA strategies within the broader context of CRISPR gene editing research.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) system functions as an adaptive immune mechanism in bacteria and archaea, but has been repurposed as a powerful genome-editing tool in molecular biology [36]. The CRISPR-Cas9 system fundamentally consists of the Cas9 nuclease and a guide RNA (gRNA) that directs it to a specific DNA sequence [37]. The simplicity of this system, compared to previous gene-editing technologies like TALENs or ZFNs, has accelerated its adoption across research and therapeutic applications [36].
The gRNA is a synthetic fusion of two natural RNA components: the CRISPR RNA (crRNA) containing the 20-nucleotide spacer sequence that determines targeting specificity through Watson-Crick base pairing, and the trans-activating crRNA (tracrRNA) which provides a structural scaffold for Cas9 binding [36]. When these two elements are combined into a single guide RNA (sgRNA), they form a functional complex that directs Cas9 to create double-strand breaks in target DNA [36]. This break occurs approximately 3 base pairs upstream of a Protospacer Adjacent Motif (PAM), a short DNA sequence adjacent to the target site that is essential for recognition by the Cas nuclease [37]. For the most commonly used Cas9 from Streptococcus pyogenes (SpCas9), the PAM sequence is 5'-NGG-3', where "N" represents any nucleotide [38].
The fundamental goal in gRNA design is to select a 20-nucleotide target sequence immediately upstream of a PAM site that is unique within the genome and possesses optimal characteristics for high editing efficiency and minimal off-target activity [37]. The design process must also consider the specific application (e.g., gene knockout, knock-in, activation, or inhibition) as optimal parameters differ based on experimental goals [39] [40]. As CRISPR technology continues to evolve, proper gRNA design remains the critical determinant of experimental success across basic research and therapeutic development.
Key Parameters for Effective gRNA Design
On-Target Efficiency
On-target efficiency refers to the predicted editing effectiveness of a gRNA at its intended target site. Various algorithms have been developed to predict gRNA on-target efficiency based on large-scale experimental data. The following table summarizes the major scoring methods and their applications:
Table 1: Key Scoring Algorithms for gRNA On-Target Efficiency
| Algorithm | Development Reference | Basis of Prediction | Application in Tools |
|---|---|---|---|
| Rule Set 1 | Doench et al. 2014 [37] | Knock-out efficiency data of 1,841 sgRNAs; considers 30nt sequence including bind area, PAM, and nearby sequences | CHOPCHOP [37] |
| Rule Set 2 | Doench et al. 2016 [37] | Based on efficiency data of 4,390 sgRNAs; uses gradient-boosted regression trees | CHOPCHOP, CRISPOR [37] |
| Rule Set 3 | DeWeirdt et al. 2022 [37] | Trained on 47k gRNAs from 7 datasets; incorporates tracrRNA considerations | GenScript, CRISPick [37] |
| CRISPRscan | Moreno-Mateos et al. 2015 [37] | Predictive model based on 1,280 gRNAs validated in vivo in zebra fish | CHOPCHOP, CRISPOR [37] |
| Lindel | Chen et al. 2019 [37] | Logistic regression model predicting indels from Cas9 cleavage; analyzes ∼1.16 million mutation events | CRISPOR [37] |
These scoring systems evaluate sequence features that influence efficiency, including nucleotide composition (e.g., guanine-rich sequences in the 5' end often perform better), position-specific determinants, and the sequence context surrounding the target site [37]. The Doench 2016 efficiency score is typically expressed as a percentile, where a score of "70%" indicates that 70% of mammalian guides have equal or lower efficiency [41].
Off-Target Specificity
Off-target effects occur when the gRNA directs Cas9 to edit unintended genomic locations with sequence similarity to the target site, potentially leading to unintended consequences in research and therapeutic contexts [37]. Specificity is a critical concern, particularly for clinical applications. The major assessment methods include:
Homology Analysis: Identifies sequences similar to the gRNA across the genome, with fewer mismatches indicating higher off-target potential. Sequences with only one mismatch pose high off-target risk, while those with zero mismatches should be eliminated from consideration [37]. Positional weighting accounts for the fact that mismatches closer to the PAM sequence typically reduce off-target effects more significantly than those farther away [37].
MIT Specificity Score: Also known as the Hsu-Zhang score, this method summarizes all potential off-targets into a single number from 0-100, with higher numbers indicating fewer expected off-target effects [37] [41]. Guides with MIT specificity scores >50 are generally recommended [41].
Cutting Frequency Determination (CFD): Developed in Doench's 2016 study, CFD uses a scoring matrix based on the activity of 28,000 gRNAs with single variations [37]. CFD scores below 0.05 (or 0.023 in some implementations) indicate low off-target risk [37].
The following table compares these off-target assessment methods:
Table 2: Comparison of Off-Target Assessment Methods
| Method | Basis | Scoring System | Threshold Guidelines |
|---|---|---|---|
| Homology Analysis | Genome-wide search for similar sequences with PAM | Number and position of mismatches | Avoid sequences with 0 mismatches; limit those with 1-3 mismatches [37] |
| MIT Specificity Score | Analysis of indel mutations in gRNA variants with 1-3 mismatches [37] | 0-100 scale (higher = better specificity) | >50 recommended [41] |
| Cutting Frequency Determination (CFD) | Activity data from 28,000 gRNAs with single variations [37] | Multiplicative matrix (scores <1) | <0.05 (or 0.023) indicates low risk [37] |
Application-Specific Design Considerations
gRNA design must be tailored to the specific experimental goal, as optimal parameters differ significantly across applications:
Gene Knockout: For CRISPR knockout (CRISPR-ko) experiments, gRNAs should target early exons common to all transcript variants, preferably in domains essential for protein function [39] [42]. Targeting regions too close to the N- or C-terminus should be avoided, as alternative start codons or non-essential protein regions may preserve function [39]. In pooled screens, using multiple gRNAs per gene (typically 3-4) improves confidence in results [42].
Knock-in Experiments: For precision editing requiring homology-directed repair (HDR), the target site must be immediately adjacent to the intended edit location, severely limiting gRNA options [39]. The repair template should ideally incorporate mutations that disrupt the PAM sequence to prevent re-cleavage after successful editing [38].
CRISPR Activation (CRISPRa) and Interference (CRISPRi): For gene regulation rather than editing, gRNAs must target specific promoter regions. CRISPRa gRNAs are most effective when located 500-50 bp upstream of the transcription start site (TSS), while CRISPRi gRNAs work best from -50 to +300 bp relative to the TSS [40].
The experimental workflow for gRNA design involves identifying the target region, locating PAM sites, designing candidate gRNAs, and evaluating them based on efficiency and specificity scores before selection and validation.
Bioinformatics Tools for gRNA Design
Comprehensive Tool Comparison
Numerous bioinformatics tools have been developed to facilitate gRNA design by incorporating the key parameters discussed above. The selection of an appropriate tool depends on the specific experimental requirements, organism, and CRISPR application. The following table summarizes major gRNA design platforms:
Table 3: Comparison of Major gRNA Design Tools
| Tool | Developer/Platform | Key Features | Scoring Algorithms | Supported Systems |
|---|---|---|---|---|
| CRISPick | Broad Institute [37] | Simple interface, provides both on-target and off-target scores | Rule Set 2/3, CFD [37] | SpCas9, other nucleases |
| CHOPCHOP | Harvard University [37] [12] | Visual off-target representations, batch processing, multiple organisms | Rule Set 1/2, CRISPRscan [37] | Various CRISPR-Cas systems |
| CRISPOR | Tefor.net [37] | Detailed off-target analysis with position-specific mismatch scoring | Rule Set 2, CRISPRscan, Lindel [37] | SpCas9, with restriction enzyme info |
| GenScript sgRNA | GenScript [37] | Integrated ordering capability, transcript visualization | Rule Set 3, CFD [37] | SpCas9, AsCas12a (coming soon) |
| CRISPRware | UC Santa Cruz [43] | Targets unannotated genomic regions, integration with Genome Browser | Custom algorithm for novel regions | Multiple CRISPR systems |
| Synthego CRISPR | Synthego [39] | Supports 120,000 genomes, 9,000 species, fast design | Doench rules for on/off-target [39] | Focus on knockouts |
These tools typically allow researchers to input a target gene or genomic region, then generate and rank potential gRNAs based on multiple parameters. Most tools filter out or flag gRNAs with potential sequence-specific off-targets and check for appropriate PAM sequences adjacent to the gRNA [40]. Advanced tools like CRISPOR provide detailed experimental considerations such as restriction enzyme sites for cloning [37].
Emerging Tools and AI Approaches
The field of gRNA design continues to evolve with emerging technologies. CRISPRware, a recently developed tool, addresses the challenge of designing gRNAs for unannotated or less-characterized genomic regions, such as those encoding small functional peptides [43]. Integrated directly into the widely used UCSC Genome Browser, CRISPRware democratizes gRNA design by making it accessible to researchers without deep bioinformatics expertise [43].
Artificial intelligence approaches are also revolutionizing gRNA and CRISPR system design. A 2025 study demonstrated the use of large language models trained on biological diversity to generate novel CRISPR-Cas proteins [44]. These AI-designed editors, such as OpenCRISPR-1, show comparable or improved activity and specificity relative to SpCas9 while being highly divergent in sequence [44]. This AI-driven approach represents a paradigm shift from mining natural systems to computationally generating optimized editing tools.
Experimental Protocols and Validation
gRNA Design Workflow for Gene Knockout
A robust protocol for designing gRNAs for gene knockout experiments involves systematic steps from target selection to validation:
Target Region Identification: Select exons common to all transcript variants of the target gene, prioritizing protein domains essential for function. Avoid regions close to the N- or C-terminus where editing may not completely disrupt gene function [39].
PAM Site Localization: Using tools like SnapGene or online design platforms, identify all NGG PAM sequences (for SpCas9) in the target region on both DNA strands [38].
gRNA Generation and Scoring: For each PAM site, extract the 20-nucleotide sequence immediately upstream. Input these candidate gRNAs into design tools like CRISPick or CHOPCHOP to obtain efficiency scores (e.g., Rule Set 2/3) and specificity metrics (e.g., CFD scores) [37].
Specificity Optimization: Filter candidates with high off-target potential (CFD > 0.05 or MIT specificity < 50) [37] [41]. Prioritize gRNAs with minimal homology to other genomic regions, especially those with fewer than 3 mismatches.
Final Selection: Choose 3-4 top-ranked gRNAs with high on-target efficiency and low off-target risk for experimental validation [42]. For critical applications, consider gRNAs with balanced GC content (30-70%) to enhance stability and minimize secondary structure formation.
Validation Methodologies
Experimental validation of gRNA efficacy and specificity is essential before proceeding with full-scale experiments:
In Vitro Cleavage Assays: Test gRNA activity using purified Cas9 protein and PCR-amplified target sequences. Measure cleavage efficiency through gel electrophoresis or fluorescent reporter systems [37].
Cell-Based Validation: Transfert candidate gRNAs with Cas9 into relevant cell lines and assess editing efficiency at the target locus 48-72 hours post-transfection using mismatch detection assays (e.g., T7E1 or Surveyor assays) or tracking of indels by decomposition (TIDE) [42].
Off-Target Assessment: Utilize genome-wide methods like CIRCLE-seq or GUIDE-seq to empirically identify off-target sites, particularly for therapeutic applications [40]. For most research applications, computational prediction with tools like Cas-OFFinder provides sufficient off-target characterization [12].
Functional Validation: For knockout experiments, verify loss of protein expression via Western blot or immunofluorescence. For knock-in approaches, confirm precise editing through PCR screening and Sanger sequencing [38].
The following diagram illustrates the key components and workflow in a typical CRISPR-Cas9 experiment:
The Scientist's Toolkit: Essential Research Reagents
Successful CRISPR experiments require carefully selected reagents and components. The following table outlines essential materials and their functions:
Table 4: Essential Research Reagents for CRISPR Experiments
| Reagent/Material | Function | Considerations |
|---|---|---|
| Cas9 Nuclease | Creates double-strand breaks at target DNA sites | Choose between wildtype, nickase, or dead Cas9 variants based on application [38] |
| gRNA Expression Vector | Plasmid for in vivo expression of gRNA | Typically uses U6 or T7 promoters; includes scaffold sequence [38] [42] |
| Repair Template (for HDR) | DNA template for precise edits via homologous recombination | For edits <200nt: ssODN with ~80-200nt homology arms; for larger edits: double-stranded DNA with 500-800nt arms [38] |
| Delivery Vehicle | Introduces CRISPR components into cells | Lentivirus, adenovirus, nanoparticles, or electroporation; choice depends on cell type and application [42] |
| Validation Primers | PCR amplification of target locus for editing assessment | Design primers ~100-200bp flanking cut site; avoid repetitive regions [38] |
| Selection Markers | Enrichment for successfully transfected cells | Antibiotic resistance genes (puromycin, blasticidin) or fluorescent reporters (GFP, RFP) [42] |
Additional specialized reagents include Cas9-positive control gRNAs targeting essential genes, negative control non-targeting gRNAs with no genomic matches, and quality control enzymes for verifying reagent integrity [42]. For therapeutic applications, high-purity, endotoxin-free preparation of all components is essential.
Effective gRNA design remains a cornerstone of successful CRISPR research, balancing multiple parameters including on-target efficiency, off-target specificity, and application-specific requirements. The development of sophisticated bioinformatics tools has dramatically improved our ability to predict gRNA performance, yet experimental validation remains essential. As the field advances, several emerging trends are shaping the future of gRNA design and CRISPR technology.
The integration of artificial intelligence and machine learning approaches is revolutionizing both gRNA design and the development of novel editing systems. AI-designed editors like OpenCRISPR-1 demonstrate that computational approaches can generate highly functional proteins divergent from natural sequences [44]. Similarly, tools like CRISPRware are making precision genome editing accessible to researchers without deep computational expertise by integrating with familiar platforms like the UCSC Genome Browser [43].
As CRISPR applications expand beyond basic research into therapeutic development, gRNA design considerations must incorporate additional factors such as delivery constraints, immune responses, and long-term safety. The successful clinical application of CRISPR for conditions like sickle cell anemia demonstrates the transformative potential of this technology when coupled with careful design principles. By adhering to rigorous gRNA design parameters and validation protocols, researchers can harness the full potential of CRISPR technology to advance both basic science and therapeutic development.
The transformative potential of CRISPR-based genome editing is fundamentally constrained by a central challenge: the efficient and safe delivery of its molecular components into target cells. The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) system, functioning as a programmable genomic scalpel, requires precise intracellular delivery to exert its therapeutic effects. Delivery systems serve as the critical transport vehicles that navigate biological barriers to place CRISPR machinery at its site of action. These systems are broadly categorized into viral vectors, which leverage evolved biological mechanisms for gene transfer, and non-viral methods, including synthetic nanoparticles and pre-assembled ribonucleoprotein (RNP) complexes. The selection of an appropriate delivery platform directly dictates the efficacy, specificity, and safety of CRISPR interventions, making it a pivotal consideration in therapeutic development [45] [46].
The cargo for CRISPR-mediated editing can be delivered in three primary forms: DNA (as plasmids encoding Cas nuclease and guide RNA), RNA (mRNA for Cas protein and the guide RNA), or as a pre-complexed ribonucleoprotein (RNP) complex (Cas protein bound to guide RNA). Each format presents distinct advantages and challenges related to editing efficiency, duration of activity, immunogenicity, and risk of off-target effects [45]. This technical guide examines the core principles, comparative performance, and experimental protocols for the three predominant delivery platforms—viral vectors, nanoparticles, and RNP complexes—within the broader context of CRISPR gene editing research for therapeutic applications.
CRISPR Cargo Types and Delivery Vehicles
The functional core of the CRISPR system consists of two components: the Cas nuclease, which cuts the DNA, and the guide RNA (gRNA), which directs the nuclease to a specific genomic locus. The format in which these components are delivered significantly impacts the editing outcome. The three primary cargo types are:
- Plasmid DNA (pDNA): Circular DNA vectors encoding genes for both the Cas protein and the gRNA. Upon cellular entry, the host machinery must transcribe and translate these genes, leading to delayed editing onset and prolonged nuclease expression that increases off-target risks [47] [45].
- Messenger RNA (mRNA) and gRNA: Synthetic mRNA encoding the Cas protein is co-delivered with the in vitro transcribed gRNA. Translation of the mRNA into functional Cas protein occurs within the cytoplasm, resulting in faster editing onset than pDNA. However, the RNA molecules are inherently unstable and can trigger innate immune responses [45].
- Ribonucleoprotein (RNP) Complexes: The Cas protein is pre-complexed with the gRNA in vitro before delivery. This complex is immediately active upon reaching the nucleus, leading to rapid editing with the shortest window of nuclease activity. This transient presence minimizes off-target effects and avoids the risk of genomic integration, making RNPs a favored option for precise ex vivo editing [47] [45].
The choice of cargo is inextricably linked to the selection of the delivery vehicle, as summarized in Table 1.
Table 1: Comparison of Primary CRISPR Delivery Vehicles and Compatible Cargos
| Delivery Vehicle | Primary Cargo Compatibility | Key Advantage | Primary Limitation | Typical Application |
|---|---|---|---|---|
| Adeno-Associated Virus (AAV) | DNA, gRNA (size-constrained) | Favorable safety profile, high tissue specificity [48] | Limited packaging capacity (~4.7 kb) [48] [45] | In vivo gene therapy |
| Lentivirus (LV) | DNA | High efficiency, stable integration, large cargo capacity [45] | Risk of insertional mutagenesis [45] [49] | Ex vivo cell engineering |
| Lipid Nanoparticle (LNP) | RNA, RNP | Minimal immunogenicity, targeted delivery (e.g., liver) [26] [45] | Endosomal entrapment, complex manufacturing [45] | In vivo & ex vivo delivery |
| Electroporation | RNP, RNA | High efficiency for hard-to-transfect cells [46] | Primarily restricted to ex vivo use [46] | Ex vivo editing of immune cells |
| Virus-Like Particle (VLP) | RNP, Protein | High transduction efficiency, no viral genome [45] | Manufacturing challenges, cargo size limits [45] | Emerging in vivo delivery |
The following diagram illustrates the logical workflow for selecting an appropriate delivery system based on the research or therapeutic objective.
Viral Vector Delivery Systems
Viral vectors are engineered viruses that have been stripped of their pathogenic components but retain their natural ability to efficiently transduce cells. They are among the most common vehicles for delivering CRISPR components in vivo and ex vivo.
Recombinant Adeno-Associated Virus (rAAV)
rAAV vectors are derived from non-pathogenic parvoviruses and are a leading platform for in vivo gene therapy, including CRISPR delivery. Their favorable safety profile, low immunogenicity, and capacity for long-term transgene expression make them particularly attractive [48] [45]. The primary constraint of rAAV is its limited packaging capacity of approximately 4.7 kb, which is insufficient for the coding sequence of the commonly used Streptococcus pyogenes Cas9 (SpCas9), which is about 4.2 kb, plus the necessary regulatory elements and gRNA expression cassette [48] [45].
Innovative Strategies to Overcome AAV Size Limitations:
- Compact Cas Orthologs: Utilizing naturally smaller Cas proteins, such as those from Staphylococcus aureus (SaCas9, ~3.2 kb) or Campylobacter jejuni (CjCas9), which can be packaged into a single AAV vector alongside their gRNA [48].
- Dual AAV Systems: The CRISPR components are split between two separate AAV vectors. One common strategy involves packaging the Cas9 nuclease in one vector and the gRNA in another. For more complex editors like base editors or prime editors, the protein can be split into two halves, each delivered by a separate vector, which reconstitute inside the target cell [48].
- Trans-splicing AAV Vectors: Employing inteins (protein-splicing elements) where the Cas protein is split and each part is delivered by a separate AAV. Following co-transduction, the inteins facilitate protein trans-splicing to form a fully functional nuclease [48].
Lentiviral and Adenoviral Vectors
- Lentiviral Vectors (LVs): LVs are RNA viruses capable of integrating their cargo into the host genome, enabling long-term, stable expression of CRISPR components. This is advantageous for ex vivo applications like engineering chimeric antigen receptor (CAR) T-cells [45] [50]. However, the risk of insertional mutagenesis and persistent expression of Cas9, which increases the potential for off-target edits, are significant safety concerns that limit their use for in vivo therapeutic editing [45] [49].
- Adenoviral Vectors (AdVs): AdVs are DNA viruses with a very large packaging capacity (up to 36 kb), easily accommodating large CRISPR cassettes. They do not integrate into the host genome, offering a transient expression profile that is safer than LVs. However, they often trigger strong host immune responses, which can lead to inflammation and rapid clearance of the transduced cells, limiting their re-administration [45].
Table 2: Detailed Comparison of Viral Vectors for CRISPR Delivery
| Vector Property | Adeno-Associated Virus (AAV) | Lentivirus (LV) | Adenovirus (AdV) |
|---|---|---|---|
| Packaging Capacity | ~4.7 kb [45] | ~8 kb [45] | Up to ~36 kb [45] |
| Genomic Integration | Non-integrating (primarily episomal) [45] | Integrating [45] | Non-integrating [45] |
| Expression Duration | Long-term (months to years) [48] | Long-term (stable) [45] | Transient (weeks) [45] |
| Immunogenicity | Low to Moderate [45] | Moderate | High [45] |
| Production Titer | High | Moderate | High |
| Key Clinical Safety Concern | Pre-existing immunity, cellular immune response | Insertional mutagenesis, genotoxicity | Inflammatory and immune responses |
Non-Viral Delivery: Nanoparticles and RNP Complexes
Non-viral delivery methods have gained significant traction due to their improved safety profiles, reduced immunogenicity, and capacity for transient delivery, which aligns with the goal of minimizing off-target effects in CRISPR therapy.
Lipid Nanoparticles (LNPs)
LNPs are synthetic, spherical vesicles composed of ionizable lipids, phospholipids, cholesterol, and lipid-anchored polyethylene glycol (PEG). They encapsulate and protect nucleic acid or protein cargo and facilitate cellular delivery through endocytosis. A landmark achievement for LNP technology was its successful use in delivering mRNA COVID-19 vaccines, which paved the way for its application in CRISPR delivery [45].
Mechanism and Workflow for LNP-mediated CRISPR Delivery: LNPs are particularly effective for delivering Cas9 mRNA and sgRNA. The ionizable lipids within LNPs are critical for endosomal escape; they become protonated in the acidic environment of the endosome, disrupting the endosomal membrane and releasing the cargo into the cytoplasm. The mRNA is then translated into the Cas protein, which complexes with the sgRNA, and the RNP enters the nucleus to perform editing [26] [45]. Recent advances include Selective Organ Targeting (SORT) LNPs, where the addition of a supplementary SORT molecule enables precise targeting of tissues beyond the liver, such as the lungs and spleen [45].
Ribonucleoprotein (RNP) Complexes
RNP complexes represent the most direct method for delivering CRISPR activity. They are formed by pre-incubating purified Cas9 protein with in vitro transcribed or synthetic sgRNA. This pre-complexed unit is then delivered into cells, most commonly via electroporation for ex vivo applications or using specialized nanoparticles for in vivo use [47].
Key Advantages of RNP Delivery:
- Rapid Editing and High Precision: As RNPs are active immediately upon nuclear entry and are quickly degraded by cellular proteases, the window for genome editing is short (typically 12-24 hours). This drastically reduces the probability of off-target editing [47].
- Reduced Cytotoxicity and Immune Recognition: Unlike plasmid DNA or viral vectors, RNPs do not require transcription or translation, which reduces cellular stress. Furthermore, they bypass the potential for anti-Cas immune responses triggered by prolonged expression from DNA templates [47].
- High Editing Efficiency: Multiple studies have demonstrated that RNP delivery, especially via electroporation, achieves higher editing efficiencies in hard-to-transfect primary cells, including hematopoietic stem cells and T-cells, compared to plasmid-based methods [47].
The following diagram outlines a standard experimental workflow for achieving gene knockout via RNP delivery and electroporation.
Table 3: Quantitative Comparison of CRISPR Cargo Formats (Based on Ex Vivo Data)
| Performance Metric | Plasmid DNA | mRNA + gRNA | RNP Complex |
|---|---|---|---|
| Time to Onset of Editing | ~24-48 hours (requires transcription/translation) [47] | ~4-12 hours (requires translation only) [45] | Immediate (0-4 hours) [47] |
| Duration of Nuclease Activity | Prolonged (days to weeks) [47] | Moderate (days) [45] | Short (12-24 hours) [47] |
| Relative Off-Target Effect | High [47] | Moderate | Low [47] |
| Editing Efficiency in Primary Cells | Variable, often low [47] | Good | High (e.g., >70% in immortalized lines) [47] |
| Relative Cytotoxicity | High (especially at high concentrations) [47] | Moderate | Low [47] |
The Scientist's Toolkit: Research Reagent Solutions
Successful execution of CRISPR delivery experiments requires a suite of high-quality reagents and materials. The following table details essential components for a research pipeline focusing on RNP and nanoparticle-based delivery.
Table 4: Essential Research Reagents for CRISPR Delivery Experiments
| Reagent / Material | Function and Critical Features | Example Application / Note |
|---|---|---|
| High-Purity Cas9 Nuclease | Recombinant, endotoxin-free Cas9 protein for RNP formation. High purity is critical for efficiency and low cytotoxicity. | For RNP assembly; available from various commercial suppliers in research-grade and GMP-grade for therapeutics [51] [47]. |
| Synthetic sgRNA | Chemically modified, high-purity single-guide RNA. Synthetic guides offer higher consistency and can be modified to enhance stability and reduce immunogenicity [47]. | Superior to in vitro transcribed (IVT) sgRNA due to reduced innate immune activation and higher editing efficiency [47]. |
| Electroporation System | Instrument that applies electrical pulses to temporarily permeabilize cell membranes, allowing RNP entry. | Systems like the Neon (Thermo Fisher) or 4D-Nucleofector (Lonza) are optimized for different cell types. Critical for ex vivo RNP delivery [46]. |
| GMP-Grade Manufacturing | Reagents produced under Current Good Manufacturing Practice guidelines. Ensures identity, purity, potency, and safety for clinical applications [51]. | Required for clinical trial material. A key challenge is procuring true GMP sgRNA and nuclease, not just "GMP-like" [51]. |
| Lipid Nanoparticles (LNPs) | Pre-formulated kits or custom LNPs for encapsulating mRNA or RNP cargo. | Used for in vivo delivery or for transfecting sensitive cell types in vitro. Compositions can be tuned for organ-specific targeting (e.g., SORT LNPs) [26] [45]. |
The development of effective CRISPR-based therapeutics is a journey that is critically dependent on overcoming the delivery challenge. Viral vectors, nanoparticles, and RNP complexes each offer a distinct set of advantages and trade-offs. Viral vectors like AAV provide efficient in vivo delivery and sustained expression but are hampered by packaging constraints and immunogenicity. Non-viral strategies, particularly LNPs, have emerged as powerful and versatile platforms for in vivo delivery of mRNA and RNP cargo. For ex vivo applications requiring maximum precision and safety, electroporation of RNP complexes is often the gold standard, enabling rapid, high-efficiency editing with minimal off-target effects.
The future of CRISPR delivery lies in the continued refinement of these platforms—engineering novel AAV serotypes and capsids with enhanced tropism and reduced immunogenicity, developing next-generation LNPs with improved tissue specificity and delivery efficiency, and optimizing RNP formulations for in vivo use. The choice of delivery system is not one-size-fits-all but must be strategically aligned with the specific therapeutic target, the desired duration of editing, and the requisite safety profile. As these technologies mature, they will undoubtedly unlock the full potential of CRISPR gene editing, transforming promising research into routine clinical reality.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) proteins constitute an adaptive immune system in prokaryotes that has been repurposed as a revolutionary genome-editing tool [52]. The system functions as a precise genetic scissor, where a guide RNA (gRNA) directs the Cas nuclease to a specific DNA sequence, enabling targeted genetic modifications [53] [52]. This programmable nature allows researchers to systematically perturb genes and investigate their functional outcomes on a large scale, an approach central to modern functional genomics [54].
Functional genomics aims to elucidate the roles and interactions of genes and genetic elements. CRISPR-based screens represent a powerful "perturbomics" approach, which involves systematically modulating gene function and analyzing the resulting phenotypic changes to annotate gene function [54]. Compared to previous technologies like RNA interference (RNAi), CRISPR screens offer greater precision, more consistent results, and fewer off-target effects, making them the preferred method for large-scale genetic interrogation [55] [42]. The core advantage lies in CRISPR's simplicity: redirecting the system to a new genomic target requires only a change in the gRNA sequence, making genome editing akin to programming a navigation system [52]. This flexibility has enabled the development of diverse screening modalities, including knockout, interference, and activation screens, to comprehensively dissect gene function.
CRISPR Screening Approaches and Experimental Design
Large-scale CRISPR screens can be implemented in two primary formats, each with distinct methodologies, equipment requirements, and compatible assay types [55].
Pooled vs. Arrayed Screening Formats
In a pooled screen, a heterogeneous library of viral vectors, each carrying a unique gRNA, is transduced into a single population of Cas9-expressing cells [55] [42]. This creates a complex mixture of mutant cells that are cultured together and subjected to a selective pressure, such as drug treatment or nutrient deprivation. The resulting phenotypes are typically "binary," meaning cells are separated based on survival or fluorescence-activated cell sorting (FACS) [55]. After selection, the relative abundance of each gRNA is quantified via next-generation sequencing (NGS) to identify genes whose perturbation causes a growth advantage or disadvantage [56] [42].
In an arrayed screen, individual gRNAs or gRNA combinations are delivered separately, each to a distinct well in a multiwell plate [55]. This format allows for the assessment of more complex, multiparametric phenotypes using high-content imaging and other assays that measure multiple cellular parameters simultaneously [55]. Since each perturbation is physically isolated, there is no need for sequencing-based deconvolution to link a phenotype to its genetic cause.
Table 1: Comparison of Pooled and Arrayed CRISPR Screening Formats
| Feature | Pooled Screen | Arrayed Screen |
|---|---|---|
| Library Delivery | Lentiviral transduction of mixed gRNA library into a single cell population [55] | Individual gRNAs delivered separately to wells of a multiplate (transfection/transduction) [55] |
| Compatible Assays | Binary assays (e.g., viability, FACS sorting) [55] | Binary and multiparametric assays (e.g., high-content imaging) [55] |
| Phenotype Genotype Linking | Requires NGS and bioinformatic deconvolution [55] | Direct, as each well contains a known perturbation [55] |
| Throughput | High, suitable for genome-wide screens [42] | Lower, often used for focused validation or complex assays [55] |
| Cost & Infrastructure | Lower cost for large libraries; requires NGS infrastructure [55] | Higher cost for large libraries; requires robotics for high-throughput handling [55] |
CRISPR Toolbox: Knockout, Interference, and Activation
The core CRISPR-Cas9 system can be engineered to achieve different types of genetic perturbations, enabling both loss-of-function and gain-of-function studies [54].
- CRISPR Knockout (KO): The native Cas9 nuclease introduces double-strand breaks in the DNA target. When repaired by error-prone non-homologous end joining (NHEJ), this leads to small insertions or deletions (indels) that often disrupt the gene's reading frame, resulting in a non-functional protein [42].
- CRISPR Interference (CRISPRi): A catalytically "dead" Cas9 (dCas9) is fused to a transcriptional repressor domain like the Krüppel associated box (KRAB) [56] [54]. This complex binds to the promoter region of a target gene without cutting the DNA and blocks transcription, effectively knocking down gene expression [54].
- CRISPR Activation (CRISPRa): The dCas9 is fused to a transcriptional activator domain, such as VP64-p65-Rta (VPR) [56] [54]. When targeted to a gene's promoter, this complex recruits the cellular transcription machinery to enhance gene expression [54].
CRISPRi and CRISPRa offer reversible and tunable control over gene expression without altering the underlying DNA sequence, making them suitable for studying essential genes and non-coding genomic elements [56] [54].
Experimental Protocol for a Pooled CRISPR-KO Screen
The following section details a standard workflow for performing a pooled CRISPR knockout screen, from library design to hit validation [42].
sgRNA Library and Cell Line Preparation
The first critical step is selecting and cloning a single guide RNA (sgRNA) library. For a genome-wide screen, established libraries like Brunello (which targets ~19,000 human genes with 4 sgRNAs per gene) are available from repositories like Addgene [42]. The library should include both non-targeting control sgRNAs (to establish a neutral baseline) and sgRNAs targeting essential genes (as positive controls for depletion) [42].
The chosen cell model must express the Cas9 nuclease stably and uniformly. This can be achieved by using a lentiviral vector to generate a clonal or polyclonal cell line with stable Cas9 integration [42]. The Cas9-expressing cells are then transduced with the pooled sgRNA lentiviral library at a low Multiplicity of Infection (MOI) to ensure most cells receive only one sgRNA. A key consideration is maintaining sufficient library representation; typically, a coverage of 500-1000 cells per sgRNA is recommended to prevent stochastic loss of specific guides [56] [42]. Transduced cells are selected using an antibiotic like puromycin.
Phenotypic Selection and Next-Generation Sequencing
After selection, a sample of cells is harvested to represent the baseline population ("Time Point T0"). The remaining cells are subjected to the experimental condition of interest, such as a drug treatment, for a defined period [56] [54]. Subsequently, a second sample is harvested ("Time Point T1") [56].
Genomic DNA (gDNA) is isolated from both the T0 and T1 populations. The sgRNA sequences are amplified from the gDNA via PCR and analyzed using NGS [42]. The read counts for each sgRNA in the T1 sample are compared to its counts in the T0 baseline. sgRNAs that are significantly enriched or depleted in the T1 population indicate that the knockout of the corresponding gene confers a selective advantage or disadvantage, respectively, under the experimental condition [56] [42].
Data Analysis and Hit Validation
Bioinformatic pipelines are used to normalize sequencing read counts and calculate statistical significance for each gene, typically aggregating the effects of all its targeting sgRNAs [56]. Genes that rank as top hits in the primary screen require validation. This involves using individual sgRNAs (rather than a pool) to recreate the gene knockout in a fresh experiment and confirm the phenotype [56]. Further validation can include orthogonal methods, such as CRISPRi or RNAi, to rule off-target effects [55].
Advanced Applications and Technical Considerations
CRISPR screening technology continues to evolve, enabling more sophisticated and physiologically relevant investigations.
Enhanced Physiological Relevance with Organoids
A significant advancement is the application of CRISPR screens in primary human 3D organoids [56]. Organoids are in vitro cultures that preserve tissue architecture, stem cell activity, and genomic alterations of primary tissues, offering a more physiologically relevant model than conventional 2D cell lines [56]. A 2025 study demonstrated the feasibility of large-scale CRISPR knockout, interference, and activation screens in human gastric organoids to identify genes modulating response to the chemotherapy drug cisplatin [56]. This approach allows for the systematic dissection of gene-drug interactions in a model that closely mimics human tissue complexity [56].
Single-Cell Resolution with CRISPR Screens
Combining pooled CRISPR screening with single-cell RNA sequencing (scRNA-seq) represents a powerful technical leap [56] [54]. This method, known as single-cell CRISPR screen, allows for the simultaneous quantification of sgRNAs and the whole transcriptome from thousands of individual cells [56]. This enables researchers to not only identify which genes are essential for a phenotype but also to resolve how specific genetic perturbations alter gene expression networks and cell states at a single-cell resolution [56] [54].
Editing Beyond Knockouts
The CRISPR toolbox has expanded beyond simple knockouts. Base editing and prime editing technologies enable precise nucleotide changes without creating double-strand breaks, allowing for the functional screening of single-nucleotide variants [57] [54]. Furthermore, CRISPR-Cas13 systems target RNA instead of DNA, enabling high-throughput knockdown screens at the transcript level [54].
Table 2: Key Research Reagents and Tools for CRISPR Screening
| Reagent/Tool | Function | Examples & Notes |
|---|---|---|
| Cas9 Nuclease | Introduces double-strand breaks for gene knockout. | Streptococcus pyogenes Cas9 (SpCas9) is most common [42]. |
| dCas9 Effectors | Engineered for transcriptional modulation without DNA cleavage. | dCas9-KRAB (for CRISPRi), dCas9-VPR (for CRISPRa) [56] [54]. |
| sgRNA Library | Pooled collection of guides targeting genes of interest. | Genome-wide (e.g., Brunello), targeted (e.g., kinome). Available from Addgene [42]. |
| Lentiviral Vectors | Efficient delivery and stable integration of sgRNAs into cells. | Allows for selection with antibiotics (e.g., puromycin) [42]. |
| Nuclease-Inactive Cell Line | Stably expresses Cas9/dCas9 uniformly. | Improves editing efficiency and screen consistency [42]. |
| Analysis Software | Analyzes editing efficiency and indel profiles from sequencing data. | ICE (Inference of CRISPR Edits) for Sanger data; MAGeCK for NGS screen data [58]. |
CRISPR-based screens have firmly established themselves as a cornerstone of functional genomics, providing an unparalleled and systematic method for annotating gene function and identifying novel therapeutic targets [54]. The transition from classic knockout screens in immortalized cells to more complex activation/interference screens and their application in physiologically relevant models like human organoids marks a significant maturation of the technology [56]. As CRISPR systems continue to evolve with capabilities for base editing, epigenetic modification, and single-cell analysis, the scope and precision of genetic screens will continue to expand [57] [54]. This progress promises to deepen our understanding of complex biological networks and accelerate the discovery of new treatments for a wide array of human diseases.
The advent of CRISPR-Cas9 genome editing has transformed the therapeutic landscape for monogenic diseases, providing a powerful tool for precise genetic modification. This technology functions as a programmable DNA-targeting system, utilizing a guide RNA (gRNA) to direct the Cas9 nuclease to a specific genomic locus, where it induces a double-strand break (DSB). The cell's subsequent repair of this break via endogenous DNA repair pathways enables targeted genetic alterations [3]. For genetic hematological disorders like sickle cell disease (SCD) and transfusion-dependent beta thalassemia (TDT), the therapeutic strategy primarily involves an ex vivo approach: autologous hematopoietic stem and progenitor cells (HSPCs) are harvested from the patient, genetically modified in the laboratory, and then reinfused following myeloablative conditioning [59] [3]. This process effectively creates a new, genetically corrected hematopoietic system.
Sickle cell disease is caused by an A·T to T·A transversion in the sixth codon of the β-globin gene (HBB), resulting in a glutamic acid to valine substitution (Glu6Val) and the production of pathological hemoglobin S (HbS) [59]. Beta thalassemia arises from mutations that reduce or prevent the synthesis of β-globin chains [60]. The foundational principle of leading CRISPR-based therapies for these diseases is not to correct the mutated HBB gene directly, but to disrupt a key regulatory gene, BCL11A [60] [59]. BCL11A is a transcriptional repressor of fetal hemoglobin (HbF), and its disruption reactulates HbF production. Since HbF does not interact with HbS to polymerize and can functionally substitute for deficient adult hemoglobin, its reactivation ameliorates the pathophysiology of both SCD and TDT [60] [59] [61].
Methodologies and Experimental Protocols
The development of CRISPR therapies requires a structured workflow from preclinical design to clinical application. The following protocol details the ex vivo editing process used for therapies like Casgevy.
Ex Vivo Gene Editing of Hematopoietic Stem Cells
Key Reagents and Materials:
- Mobilized Apheresis Product: Collected from the patient after administration of a mobilizing agent like plerixafor to bring CD34+ HSPCs into the peripheral blood [61].
- CRISPR Components: Cas9 nuclease (or Cas12a) and single-guide RNA (sgRNA) targeting the
BCL11Aerythroid-specific enhancer or theHBG1/HBG2promoter regions [60] [61]. - Electroporation System: For delivering CRISPR ribonucleoproteins (RNPs) into the isolated CD34+ cells.
- Cell Culture Media: Specialized media for the short-term maintenance of HSPCs during and after editing.
- Myeloablative Conditioning Drug: Typically busulfan, to create marrow space for the engraftment of edited cells [61].
Step-by-Step Protocol:
- CD34+ HSPC Collection and Isolation: Perform apheresis on the patient following mobilization with plerixafor. Isulate CD34+ cells from the apheresis product using immunomagnetic selection (e.g., CliniMACS system) [61].
- CRISPR Complex Formation and Electroporation: Form ribonucleoprotein (RNP) complexes by pre-incubating the Cas9 protein with sgRNA. Electroporate the isolated CD34+ cells with the RNP complexes to introduce the genetic modification [60].
- Quality Control and Expansion: Culture the electroporated cells for a brief period (e.g., 2-3 days) in cytokine-supplemented media. Sample the cells for quality control assays, including:
- Editing Efficiency: Assessed by next-generation sequencing (NGS) of the target locus to determine the percentage of indels.
- Viability and Potency: Confirmed via flow cytometry and colony-forming unit (CFU) assays [59].
- Myeloablative Conditioning and Reinfusion: Administer myeloablative busulfan to the patient. Once the conditioning regimen is complete, thaw and infuse the edited CD34+ cells into the patient via a central venous catheter [61].
- Engraftment and Follow-up Monitoring: Monitor the patient for neutrophil and platelet engraftment. Conduct long-term follow-up to track:
This workflow is visualized in the following diagram, which outlines the key steps from cell collection to patient monitoring.
Key Therapeutic Approaches and Quantitative Outcomes
The field has converged on several strategic approaches for SCD and TDT, each with distinct molecular mechanisms and clinical outcomes.
Table 1: Comparison of Major Gene Therapy Approaches for SCD and TDT
| Therapeutic Approach | Molecular Target & Mechanism | Editing Tool | Key Clinical/Preclinical Outcomes |
|---|---|---|---|
| BCL11A Enhancer Disruption (e.g., Casgevy) | Disrupts erythroid-specific enhancer of BCL11A, a repressor of fetal hemoglobin (HbF). Leads to HbF reactivation. |
CRISPR-Cas9 [60] | - ~80% allele editing in HSPCs.- >40% HbF in patients post-treatment.- Transfusion independence in TDT (7/7 patients).- Elimination of VOCs in SCD (no events in 3/3 patients) [60] [59]. |
| HBG Promoter Editing (e.g., Reni-cel/EDIT-301) | Uses CRISPR-Cas12a to edit promoter regions of HBG1 and HBG2 to mimic natural hereditary persistence of HbF mutations. |
CRISPR-Cas12a [61] | - ~47.7% HbF by month 4 sustained.- Normal hemoglobin levels in SCD patients by month 5.- Transfusion-free in TDT (5/5 patients) [61]. |
| HBB Gene Correction | Uses HDR with a donor template to directly correct the causative Glu6Val point mutation in the HBB gene. |
CRISPR-Cas9 + Donor Template [59] | Preclinical stage: High efficiency and reproducibility in correcting the mutation in patient-derived HSPCs in weeks [59]. |
| Base Editing for HBB | Converts the pathogenic HBB allele (GTG) into a non-pathogenic variant, Makassar β-globin (GCG), without causing DSBs. |
CRISPR Base Editor [59] | Preclinical stage: Successfully alleviated SCD in murine models with higher editing efficiency and fewer genotoxicity concerns compared to Cas9 in competitive transplants [59]. |
The core biological mechanism of the BCL11A targeting strategy is detailed in the pathway diagram below, illustrating how disruption of the enhancer leads to therapeutic HbF expression.
Advanced Technical and Research Considerations
The Scientist's Toolkit: Essential Reagents and Materials
Successful development and implementation of ex vivo gene therapies require a suite of specialized research reagents and platforms.
Table 2: Essential Research Reagent Solutions for Ex Vivo Gene Therapy
| Reagent/Material | Function | Specific Examples & Notes |
|---|---|---|
| Programmable Nuclease Systems | Induces targeted DNA breaks for gene disruption or correction. | CRISPR-Cas9: Most widely used [60]. CRISPR-Cas12a: Used in EDIT-301; different PAM requirement can offer unique targeting options [61]. Base Editors: Enable precise single-nucleotide changes without DSBs, potentially safer [59] [3]. |
| Guide RNA (gRNA) | Provides targeting specificity by complementary base pairing with the DNA locus. | Designed to target the BCL11A erythroid-specific enhancer or the HBG promoter. Must be screened for on-target efficiency and potential off-target effects [60] [3]. |
| HSPC Isolation Kits | Purifies CD34+ hematopoietic stem cells from apheresis product. | Immunomagnetic positive selection kits (e.g., CliniMACS CD34+ reagent) are critical for obtaining a pure cell population for editing [61]. |
| Electroporation Systems | Enables efficient delivery of CRISPR RNP complexes into sensitive HSPCs. | Systems like the Lonza 4D-Nucleofector are optimized for high viability and editing efficiency in primary human CD34+ cells. |
| Specialized Cell Culture Media | Supports the survival, maintenance, and minimal expansion of HSPCs during ex vivo manipulation. | Serum-free media supplemented with recombinant cytokines (e.g., SCF, TPO, FLT3-L) is essential to preserve stemness [59]. |
| Analytical Tools for QC | Measures the success and safety of the editing process. | NGS Platforms: For on-target editing efficiency and off-target profiling. Flow Cytometry: For cell viability, CD34+ purity, and HbF expression. CFU Assays: To measure progenitor cell functionality post-editing [59]. |
Innovations and Future Directions
The field is rapidly advancing beyond first-generation Cas9 therapies. Key innovations include:
- Novel Editing Platforms: Base editing and prime editing are being actively explored to create more precise genetic modifications without inducing DSBs, thereby reducing the risk of unwanted indels and improving the safety profile [59] [3]. For instance, a base editing approach successfully converted the sickle allele to a non-pathogenic Makassar β-globin variant in mouse models [59].
- Delivery Technologies: While ex vivo delivery is established for blood disorders, in vivo delivery is a major frontier. Lipid Nanoparticles (LNPs) have proven highly effective for delivering CRISPR components to the liver for other diseases, demonstrating potential for redosing, which is not feasible with viral vectors [26].
- Overcoming Biological Challenges: Recent research highlights that DNA repair pathways differ significantly between dividing cells and non-dividing cells (e.g., neurons, cardiomyocytes). One study found that Cas9-induced indels accumulate much more slowly in postmitotic neurons, and these cells utilize different DNA repair factors, which presents both challenges and opportunities for therapeutic editing in non-proliferative tissues [62]. This underscores the necessity of studying editing outcomes in clinically relevant cell types.
CRISPR-based gene therapies for sickle cell anemia and beta thalassemia represent a paradigm shift in the treatment of genetic disorders, moving from symptomatic management to a potential one-time curative treatment. The success of Casgevy and the promising clinical results of EDIT-301 validate the strategy of targeting the BCL11A enhancer or HBG promoters to reactivate fetal hemoglobin. The detailed experimental protocols for the ex vivo editing, expansion, and transplantation of HSPCs provide a roadmap for researchers and clinicians. As the field progresses, the integration of next-generation editing tools like base editors and prime editors, coupled with advanced delivery systems, promises to further enhance the efficacy, safety, and applicability of gene therapies for a broader range of genetic diseases.
The field of genome editing has evolved dramatically from early nuclease-based technologies to modern precision editing tools that can rewrite genetic information with single-base resolution. While CRISPR-Cas9 nucleases revolutionized biological research by enabling targeted DNA cleavage, their reliance on double-strand breaks (DSBs) and cellular repair mechanisms introduces significant limitations, including unpredictable insertions/deletions (indels), chromosomal rearrangements, and activation of DNA damage response pathways [63] [64]. These challenges are particularly problematic for therapeutic applications where precision is paramount. Base editing and prime editing represent two groundbreaking technologies that address these limitations by enabling precise genetic modifications without requiring DSBs. These technologies expand the capabilities of CRISPR-based editing and offer new avenues for therapeutic development, functional genomics, and agricultural improvement [65] [66].
The clinical relevance of these technologies is underscored by the recent approval of the first CRISPR-based medicine, Casgevy, for sickle cell disease and transfusion-dependent beta thalassemia [26] [66]. However, this therapy still relies on traditional CRISPR-Cas9 nuclease activity. Base editors and prime editors offer potentially safer alternatives for correcting point mutations, which account for approximately 90% of known pathogenic genetic variants [67]. As the field advances toward more precise genetic medicines, understanding the mechanisms, capabilities, and limitations of these next-generation editing tools becomes essential for researchers and drug development professionals.
Understanding Base Editing
Molecular Mechanism of Base Editors
Base editors are fusion proteins that combine a catalytically impaired Cas protein (either dead Cas9/dCas9 or nickase Cas9/nCas9) with a nucleotide deaminase enzyme to enable direct chemical conversion of one DNA base to another without generating DSBs [67] [64]. The system is guided to a specific genomic locus by a guide RNA (gRNA), where the Cas component binds to DNA and unwinds the double helix, exposing a single-stranded DNA region. The deaminase enzyme then acts on a specific base within this exposed region, catalyzing its conversion to a different base [67]. The editing outcome is determined by which deaminase is used and the subsequent cellular repair processes.
There are two primary classes of base editors: Cytosine Base Editors (CBEs) and Adenine Base Editors (ABEs). CBEs convert cytosine (C) to thymine (T), effectively producing a C•G to T•A base pair substitution [67] [64]. This conversion is typically mediated by the cytidine deaminase APOBEC1, which converts cytosine to uracil. The cell's DNA replication machinery then interprets this uracil as thymine, completing the conversion. To enhance editing efficiency, CBEs often incorporate uracil glycosylase inhibitors (UGI) that prevent cellular repair enzymes from reversing the edit [67]. The original CBE, BE3, was published by Komor et al. in 2016 and demonstrated efficient C-to-T conversions in mammalian cells [67].
ABEs perform adenine (A) to guanine (G) conversions, resulting in A•T to G•C base pair substitutions [67] [64]. Developing ABEs presented a significant challenge as no natural DNA adenosine deaminases were known. Researchers addressed this by engineering the E. coli tRNA adenosine deaminase (TadA) through directed evolution to create a version that could deaminate adenine in DNA [67] [64]. The engineered TadA variant converts adenine to inosine, which the cellular machinery interprets as guanine during DNA replication. The first ABE (ABE7.10) was reported by Gaudelli et al. in 2017 and showed high-efficiency editing at multiple genomic sites [67].
Base Editing Mechanism: CBE and ABE utilize distinct deaminase enzymes to mediate precise base conversions without double-strand breaks.
Key Considerations for Base Editing Implementation
Successful implementation of base editing requires careful consideration of several technical parameters. The editing window is a critical concept—it refers to the narrow range of bases within the protospacer region where efficient deamination can occur [67]. For most base editors, this window spans approximately 4-5 nucleotides and is positioned at a fixed distance from the protospacer adjacent motif (PAM) site [63]. The precise positioning of the target base within this window is essential for efficient editing.
Protospacer Adjacent Motif (PAM) requirements dictated by the Cas protein component can limit targetable sites [64]. The commonly used SpCas9 requires an NGG PAM sequence immediately following the target site. While this occurs frequently in many genomes, it may not be present near desired editing sites. Researchers have addressed this limitation by developing base editors that incorporate Cas proteins with alternative PAM specificities, such as SaCas9, Cas12a, and engineered SpCas9 variants like xCas9 and SpCas9-NG [64].
Bystander edits represent another important consideration [63] [64]. These occur when additional bases of the same type within the editing window are unintentionally modified along with the target base. For example, a CBE might convert multiple cytosines within the editing window to thymines, even if only one specific C-to-T conversion was desired. Strategies to minimize bystander edits include optimizing the positioning of the target base within the editing window and engineering deaminase variants with narrower activity windows [64].
Off-target effects in base editing can manifest as both DNA and RNA deamination [64]. The deaminase components, particularly APOBEC1 in CBEs, can sometimes exhibit promiscuous activity on non-targeted DNA or RNA. Extensive engineering efforts have produced high-fidelity base editor variants with reduced off-target activity through techniques like directed evolution and rational protein design [64].
Understanding Prime Editing
Molecular Mechanism of Prime Editing
Prime editing represents a more versatile precise genome editing technology that can mediate all 12 possible base-to-base conversions, as well as small insertions and deletions, without requiring DSBs or donor DNA templates [68] [63]. The system consists of two main components: (1) a prime editor protein, which is a fusion of a Cas9 nickase (nCas9) and an engineered reverse transcriptase (RT), and (2) a prime editing guide RNA (pegRNA) that specifies the target site and encodes the desired edit [68] [63].
The prime editing mechanism occurs through a multi-step process [68] [63] [69]:
- The prime editor complex binds to the target DNA site directed by the pegRNA spacer sequence.
- The nCas9 (H840A) component nicks the non-target DNA strand, liberating a 3' hydroxyl group.
- The released 3' DNA end hybridizes with the primer binding site (PBS) sequence of the pegRNA.
- The reverse transcriptase uses the 3' DNA end as a primer and the RT template (RTT) sequence of the pegRNA as a template to synthesize DNA containing the desired edit.
- The newly synthesized edited DNA flap competes with the original unedited DNA flap for incorporation into the genome.
- Cellular DNA repair mechanisms resolve the intermediate structure, ideally incorporating the edit into the genomic DNA.
The original prime editing system (PE1) has undergone multiple iterations of optimization [68] [63]. PE2 incorporated engineered reverse transcriptase mutations (D200N/L603W/T330P/T306K/W313F) that enhance editing efficiency. PE3 adds a second nicking guide RNA (ngRNA) to nick the non-edited strand, further improving editing efficiency by encouraging the cell to use the edited strand as a repair template [68] [63]. More recently, evolved prime editors including PEmax, PE6a, PE6b, and PE6c have shown substantially improved editing efficiencies through additional optimization of the RT and Cas9 components [68].
Prime Editing Mechanism: The prime editor uses a pegRNA to direct nicking and reverse transcription, enabling versatile editing without double-strand breaks.
Advanced Prime Editing Systems and Optimization
The rapid evolution of prime editing technology has produced increasingly sophisticated systems with improved efficiency and expanded capabilities. Later-generation prime editors incorporate various enhancements, including nuclear localization signals, codon optimization, and engineered reverse transcriptase domains with improved processivity and stability [68] [63]. The development of PEmax through implementation of mutations R221K and N394K in the Cas9 region, combined with codon optimization and linker replacement, significantly improved editing efficiency compared to earlier versions [68].
Recent efforts have focused on developing more compact prime editors to enhance delivery options, particularly for viral vectors with limited cargo capacity [68]. PE6 variants incorporate evolved reverse transcriptases from various sources, including E. coli (Ec48 in PE6a) and S. pombe (Tf1 in PE6b), which offer comparable editing efficiency to PEmax despite significantly smaller size [68]. For example, PE6b is approximately 33% smaller than PEmax while maintaining high editing efficiency, making it particularly valuable for therapeutic applications where delivery constraints are paramount [68].
Additional optimization strategies include the use of engineered pegRNAs (epegRNAs) with modified 3' structures that resist exonucleolytic degradation and improve stability [68] [63]. The PE7 system further enhances editing efficiency by fusing the prime editor with the La (1-194) protein, which stabilizes the pegRNA and improves editing outcomes in challenging cell types [63]. Twin prime editing systems, which use two pegRNAs to introduce edits at both strands simultaneously, enable larger insertions (up to 250 bp) and deletions (up to 10 kb) [66].
Comparative Analysis of Editing Technologies
Technical Comparison
The table below provides a comprehensive comparison of the key technical characteristics of base editing, prime editing, and traditional CRISPR-Cas9 nuclease editing:
Table 1: Comparison of Genome Editing Technologies
| Parameter | CRISPR-Cas9 Nuclease | Base Editing | Prime Editing |
|---|---|---|---|
| Core Components | Cas9 nuclease, sgRNA | deaminase-nCas9 fusion, gRNA | RT-nCas9 fusion, pegRNA |
| DNA Break Type | Double-strand break (DSB) | Single-strand break (nick) or none | Single-strand break (nick) |
| Editing Types | Indels, large deletions | C•G to T•A (CBE) or A•T to G•C (ABE) | All 12 base conversions, insertions, deletions |
| Theoretical Editing Purity | Low (mixed outcomes) | High | Very high |
| Cell Cycle Dependence | Yes (for HDR) | No | No |
| Off-target Effects | DSB-associated indels, chromosomal rearrangements | DNA/RNA deamination, bystander edits | Relatively low indels |
| Primary Applications | Gene knockouts, large deletions | Point mutation correction, stop codon introduction | Versatile precise editing |
| Key Limitations | Unpredictable indels, low HDR efficiency | Restricted to specific transitions, bystander edits | Lower efficiency, complex pegRNA design |
Editing Outcomes and Purity
A critical advantage of both base editing and prime editing over traditional nuclease-based approaches is the significantly higher purity of desired editing outcomes. CRISPR-Cas9 nucleases produce a complex mixture of editing products, with desired HDR-mediated corrections typically occurring in less than 10% of treated cells, while the majority of outcomes are indels resulting from NHEJ repair [66]. In contrast, base editors can achieve desired base conversions in 50-80% of alleles with minimal indel formation (typically <1-5%) [64]. Prime editing generally exhibits the highest product purity, with desired edits occurring in 5-50% of alleles depending on the target site and editor version, while indel rates remain very low (often <1%) [68] [63].
This high editing purity is particularly valuable for therapeutic applications where minimizing unintended mutations is crucial for safety. However, editing efficiency remains a challenge for prime editing, especially in certain cell types and genomic contexts. Ongoing optimization efforts focus on improving prime editing efficiency through engineered components and enhanced delivery methods [68] [63].
Research Reagents and Experimental Implementation
Essential Research Reagents
Table 2: Essential Research Reagents for Base and Prime Editing
| Reagent Category | Specific Examples | Function |
|---|---|---|
| Editor Plasmids | BE4max, ABE8e, PEmax, PE6 variants | Encodes the base editor or prime editor protein components |
| Guide RNA Systems | pegRNA plasmids, epegRNA scaffolds, ngRNAs | Directs editing to specific genomic loci and encodes desired edits |
| Delivery Vehicles | AAV vectors, Lentivirus, Lipid Nanoparticles (LNPs) | Enables efficient intracellular delivery of editing components |
| Validation Tools | Sanger sequencing, Next-generation sequencing, T7E1 assay | Confirms editing efficiency and assesses off-target effects |
| Cell Culture Resources | HEK293T, HAP1, iPSCs, Primary cell media | Provides model systems for editing optimization and testing |
Experimental Workflow and Protocol
Implementing base editing or prime editing typically follows a standardized workflow:
Step 1: Target Selection and gRNA Design
- Identify the target genomic region and specific nucleotide changes required
- For base editing: Ensure the target base falls within the editing window (typically positions 4-8 in the protospacer for SpCas9-based editors) and verify PAM availability [67] [64]
- For prime editing: Design the pegRNA spacer (typically 20 nt), primer binding site (PBS, typically 10-15 nt), and reverse transcriptase template (RTT, contains desired edit) [68] [63]
- Use computational tools to predict potential off-target sites and optimize specificity
Step 2: Component Assembly
- Clone designed gRNAs or pegRNAs into appropriate expression vectors
- Combine with editor expression plasmids (e.g., BE4max for CBE, PEmax for PE)
- For prime editing, consider including nicking gRNAs (ngRNAs) for PE3 systems to enhance efficiency [68]
Step 3: Delivery to Target Cells
- Transferd dividing cell lines (HEK293T, HAP1) using lipid-based transfection
- Transduce difficult-to-transfect cells (primary cells, iPSCs) using viral vectors (AAV, lentivirus) or electroporation
- For in vivo applications, utilize lipid nanoparticles (LNPs) or viral vectors optimized for target tissue tropism [26]
Step 4: Analysis and Validation
- Extract genomic DNA 48-96 hours post-delivery
- Amplify target region by PCR and sequence using Sanger or next-generation sequencing
- Quantify editing efficiency (% of reads containing desired edit) and assess presence of bystander edits or indels
- For therapeutic applications, perform functional assays to confirm phenotypic correction
Experimental Workflow: The implementation of base and prime editing follows a systematic process from design to validation.
Clinical Applications and Future Directions
The therapeutic potential of base editing and prime editing is rapidly being realized in both preclinical studies and clinical trials. Base editors have advanced to human clinical trials for several conditions, including VERVE-101 for lowering LDL cholesterol in patients with heterozygous familial hypercholesterolemia, and BEAM-101 for sickle cell disease and beta-thalassemia [66]. These applications leverage the ability of base editors to install protective mutations or correct disease-causing point mutations with high efficiency and minimal indel formation.
Prime editing, though newer, shows tremendous promise for addressing genetic mutations that are not targetable by base editing. Recent breakthroughs include the development of the PERT (Prime Editing-mediated Readthrough of Premature Termination Codons) system, which uses a single prime editor to install a suppressor tRNA that enables readthrough of nonsense mutations across multiple different genetic diseases [70]. This approach could potentially treat approximately 30% of rare diseases caused by premature stop codons with a single therapeutic agent, dramatically expanding the addressable patient population [70].
The first personalized in vivo prime editing therapy was recently administered to an infant with CPS1 deficiency, demonstrating the rapid clinical translation of this technology [26] [71]. The therapy was developed and delivered in just six months, setting a precedent for rapid customization of gene editing treatments for individuals with rare genetic disorders [26]. This achievement highlights the potential of prime editing to address previously untreatable genetic conditions through bespoke therapies.
Future directions in the field include further optimization of editing efficiency and specificity, expansion of targeting scope through engineered Cas proteins with alternative PAM preferences, and development of more efficient delivery systems, particularly for in vivo applications [68] [63] [66]. As these technologies continue to mature, base editing and prime editing are poised to revolutionize the treatment of genetic diseases and expand the toolbox available to researchers and therapeutic developers.
Enhancing Precision and Efficiency: A Guide to Overcoming CRISPR Challenges
CRISPR-Cas9 genome editing has revolutionized biological research and therapeutic development by enabling precise DNA modifications. However, a significant challenge compromising this precision is off-target editing, which refers to the non-specific activity of the Cas nuclease at genomic sites other than the intended target, leading to unintended alterations [72]. These unintended effects occur because wild-type CRISPR systems possess a reasonable level of tolerance for mismatches between their target sequence and guide RNA (gRNA). Specifically, the wild-type Cas9 from Streptococcus pyogenes (SpCas9) can tolerate between three and five base pair mismatches, potentially creating double-stranded breaks at multiple genomic locations if they bear sufficient similarity to the intended target and contain the correct protospacer adjacent motif (PAM) sequence [72].
The ramifications of off-target editing are multifaceted. In basic research, off-target effects can confound experimental results and decrease repeatability, making phenotypic interpretation challenging [72]. In clinical applications, the risks are substantially greater; off-target edits in oncogenes or tumor suppressor genes could pose critical safety risks to patients [72]. Recent studies have revealed that the consequences extend beyond simple small insertions or deletions (indels) to include large structural variations such as chromosomal translocations, megabase-scale deletions, and chromothripsis [73]. Regulatory agencies like the FDA and EMA now require comprehensive characterization of both on-target and off-target effects for therapeutic development [73].
Predicting and Detecting Off-Target Effects
Computational Prediction Methods
The first line of defense against off-target effects begins with careful gRNA design using specialized computational tools. These tools rank all possible gRNAs for a target site based on their predicted on-target to off-target activity ratio [72]. High-ranking gRNAs typically exhibit high on-target activity with lower risk of off-target editing. Key algorithms for CRISPR off-target prediction include Cas-OFFinder and Off-Spotter, which identify potential off-target sites across the genome based on sequence similarity to the intended target, considering factors like the number and position of mismatches, and PAM compatibility [74]. Advanced approaches now incorporate machine learning frameworks, including RNN-GRU and multi-layer neural networks, with transfer learning methodologies that use cosine distance metrics to identify optimal source datasets, significantly improving prediction accuracy [75].
Experimental Detection Methods
After performing CRISPR editing, experimental validation of off-target effects is essential. Multiple methods exist with varying sensitivity, scalability, and technical requirements.
Table 1: Comparison of Methods for Detecting CRISPR Off-Target Effects
| Method | Principle | Key Applications | Advantages | Limitations |
|---|---|---|---|---|
| Candidate Site Sequencing [72] | Sequencing predicted off-target sites identified during gRNA selection | Initial safety assessment | Simple, cost-effective | Limited to predicted sites; may miss novel off-targets |
| GUIDE-Seq [72] | Integration of oligonucleotides into DSB sites followed by sequencing | Genome-wide unbiased detection | Sensitive; does not require prior knowledge of off-target sites | Complex workflow; may miss off-targets in certain genomic contexts |
| CIRCLE-Seq [72] | In vitro circularization and amplification of off-target sites | Highly sensitive pre-clinical screening | Extremely high sensitivity; works on purified genomic DNA | In vitro method may not reflect cellular context |
| DISCOVER-Seq [72] | Relies on recruitment of DNA repair factors (MRE11) to DSBs | Unbiased in-cell identification | Utilizes endogenous repair machinery; works in primary cells | Requires specific antibodies and chromatin immunoprecipitation |
| CAST-Seq [72] | Detection of chromosomal rearrangements and translocations | Safety assessment for therapeutic development | Specifically identifies structural variations | May not detect simple indels without rearrangements |
| Whole Genome Sequencing (WGS) [72] | Comprehensive sequencing of the entire genome | Gold standard for comprehensive assessment | Most complete analysis; detects all variant types | Expensive; computationally intensive; may require deep sequencing |
Each method offers distinct advantages, and researchers often employ complementary approaches for comprehensive off-target assessment [72]. For example, CAST-Seq was specifically designed to identify and quantify chromosomal rearrangements resulting from CRISPR editing, making it particularly valuable for therapeutic applications where structural variations pose significant safety concerns [72].
The diagram below illustrates a generalized workflow for off-target effect prediction and experimental validation:
Strategies to Minimize Off-Target Effects
High-Fidelity Cas Variants
Protein engineering has produced several enhanced-fidelity Cas9 variants with reduced off-target activity while maintaining robust on-target editing. These variants typically contain mutations that destabilize Cas9 binding to non-target DNA, increasing its specificity.
Table 2: High-Fidelity Cas9 Variants and Their Properties
| Variant | Key Mutations | Off-Target Reduction | On-Target Efficiency | Key Applications |
|---|---|---|---|---|
| HiFi Cas9 [74] | R691A | Significant reduction | Maintains high efficiency | Preclinical cancer models (e.g., KRAS-mutant NSCLC) |
| SpCas9-HF1 [72] | N497A, R661A, Q695A, Q926A | Enhanced specificity | Slightly reduced | General research applications |
| eSpCas9(1.1) [72] | K848A, K1003A, R1060A | Enhanced specificity | Slightly reduced | General research applications |
| OpenCRISPR-1 [44] | AI-designed (400 mutations from SpCas9) | Comparable/improved to SpCas9 | Comparable/improved to SpCas9 | Broad research and commercial use |
HiFi Cas9 has demonstrated exceptional utility in therapeutic contexts. In a 2025 study targeting KRAS driver mutations in non-small cell lung cancer (NSCLC), HiFi Cas9 enabled specific targeting of oncogenic KRASG12C and KRASG12D mutants while completely avoiding editing of wild-type KRAS (KRASWT) [74]. This specificity is crucial for therapeutic applications where targeting wild-type alleles could cause detrimental effects in healthy tissues. The researchers systematically evaluated various sgRNAs with different PAM sites and found that appropriately designed mutation-specific sgRNAs complexed with HiFi Cas9 as ribonucleoprotein particles (RNPs) could discriminate single-nucleotide differences between mutant and wild-type alleles [74].
Artificial intelligence has emerged as a powerful approach for designing novel CRISPR editors with enhanced properties. In a landmark 2025 study, researchers used large language models trained on 1 million CRISPR operons to generate artificial CRISPR-Cas proteins [44]. One resulting editor, OpenCRISPR-1, exhibits comparable or improved activity and specificity relative to SpCas9 while being 400 mutations away in sequence, demonstrating the potential of AI to bypass evolutionary constraints and generate editors with optimal properties [44].
gRNA Design and Modifications
Careful gRNA design represents the most straightforward strategy to minimize off-target effects. Several factors influence gRNA specificity:
- GC Content: Higher GC content in the gRNA sequence stabilizes the DNA:RNA duplex when the guide binds to the target, increasing on-target editing and reducing off-target binding [72].
- Guide Length: Shorter gRNAs of 20 nucleotides or less have a lower risk of off-target activity [72].
- Chemical Modifications: Adding 2'-O-methyl analogs (2'-O-Me) and 3' phosphorothioate bond (PS) modifications to synthetic gRNAs can reduce off-target edits while increasing editing efficiency at the target site [72].
- Specificity Screening: Using bioinformatics tools to screen potential gRNAs against the entire genome to select guides with minimal similar sites [72].
In proof-of-concept experiments, it's common practice to select several top-ranking gRNAs from design tools and empirically test them in relevant cell lines, as the top-ranked guide in silico may not always yield the best results in biological systems [72].
Alternative CRISPR Systems and Delivery Methods
Beyond high-fidelity Cas9 variants, several alternative strategies can reduce off-target effects:
Alternative CRISPR Systems: Cas12 and Cas13 nucleases have different off-target profiles compared to SpCas9 and may be preferable for certain applications [72].
Base and Prime Editing: These technologies can reduce the likelihood of off-target editing because they do not create double-stranded breaks in the genome [72]. Base editors use catalytically dead Cas9 (dCas9) or Cas9 nickase (nCas9) fused to deaminase enzymes to directly convert one base to another, while prime editing uses a reverse transcriptase fused to nCas9 programmed with a prime editing guide RNA (pegRNA) to copy edited sequences into the genome [76].
Delivery Method Optimization: The choice of CRISPR cargo and delivery vehicle significantly impacts off-target editing by influencing how long CRISPR components remain active in cells [72]. Short-term expression of gene editing components is highly desirable to reduce CRISPR off-target editing in clinical applications. Delivery as ribonucleoprotein (RNP) complexes rather than plasmid DNA or mRNA leads to more rapid clearance of editing activity, thereby narrowing the window for off-target effects [74].
The following diagram illustrates how high-fidelity Cas variants achieve their improved specificity:
Experimental Protocol: Assessing Specificity in Preclinical Models
This protocol outlines the methodology used in the 2025 Nature Communications study demonstrating HiFi Cas9-mediated targeting of KRAS mutations in NSCLC models [74].
Guide RNA Design and RNP Complex Formation
- Design mutation-specific sgRNAs: For KRASG12C and KRASG12D mutations, design sgRNAs complementary to the mutant sequence but containing mismatches to the wild-type sequence. Consider different PAM options (e.g., PAM1: AGG and PAM2: TGG).
- Incorporate specificity enhancements: Introduce a single mismatch adjacent to the mutated nucleotide in the KRASG12C-mutant-specific sgRNA to enhance discrimination capability.
- Form ribonucleoprotein (RNP) complexes: Complex purified HiFi Cas9 protein with synthetic sgRNAs at a molar ratio of 1:1.2 (Cas9:sgRNA) in nuclease-free buffer, incubate at room temperature for 10 minutes to allow RNP formation.
Cell Culture and Transfection
- Maintain relevant cell lines: Culture KRAS-mutant (H23, H358, A427) and KRAS wild-type (H838) NSCLC cell lines in appropriate media. Include isogenic panel of KRAS-humanized mouse embryonic fibroblast (MEF) models for controlled comparison.
- Transfert with RNP complexes: Deliver RNP complexes into cells via lipofection. For tracking transfection efficiency, use fluorescently labeled tracrRNA (CRISPR-Cas9 tracrRNA-ATTO 550) as a component of the sgRNAs.
- Include controls: Transfect parallel samples with non-HiFi Cas9 systems to compare specificity.
Specificity Assessment
T7 Endonuclease I Assay:
- Extract genomic DNA 72 hours post-transfection
- PCR-amplify target region surrounding KRAS codon 12
- Hybridize PCR products to form heteroduplexes
- Digest with T7 Endonuclease I to cleave mismatched heteroduplexes
- Analyze cleavage products by agarose gel electrophoresis
- Quantify editing efficiency using densitometric analysis
Next-Generation Sequencing (NGS):
- Amplify target region by PCR using barcoded primers
- Perform paired-end sequencing on Illumina platform
- Analyze sequencing data for indel frequencies at both mutant and wild-type alleles
- Use ICE (Inference of CRISPR Edits) tool to validate editing efficiency and characterize indel patterns
Off-Target Assessment:
- Use Cas-OFFinder and Off-Spotter algorithms to predict potential off-target sites
- Amplify top predicted off-target sites by PCR and sequence by NGS
- Compare editing frequencies at off-target sites between HiFi Cas9 and non-HiFi systems
Functional Validation
- Cell viability assays: Measure viability of KRAS-mutant versus wild-type cells after editing using MTT or similar assays.
- Downstream signaling analysis: Assess effects on KRAS signaling pathways (MAPK, PI3K) by Western blotting for phosphorylated ERK and AKT.
- Tumorigenicity assays: Evaluate transformed growth properties in soft agar and xenograft models.
Research Reagent Solutions
Table 3: Essential Research Reagents for Off-Target Assessment
| Reagent/Category | Specific Examples | Function and Application | Considerations |
|---|---|---|---|
| High-Fidelity Nucleases | HiFi Cas9 [74], SpCas9-HF1 [72], eSpCas9(1.1) [72], OpenCRISPR-1 [44] | Reduce off-target editing while maintaining on-target activity | Balance between specificity and efficiency; consider PAM requirements |
| gRNA Design Tools | CRISPOR, Cas-OFFinder [74], Off-Spotter [74] | Predict off-target sites and rank gRNAs by specificity | Use multiple algorithms for comprehensive prediction |
| Detection Kits | T7 Endonuclease I [74], GUIDE-Seq kits [72], CIRCLE-Seq kits [72] | Experimental detection and quantification of off-target effects | Match sensitivity needs with experimental context; CELL-free vs. cellular methods |
| Analysis Software | ICE (Inference of CRISPR Edits) [72] [74], TIDE [76] | Analyze sequencing data to quantify editing efficiency and characterize edits | ICE works with Sanger sequencing; NGS provides more comprehensive data |
| Delivery Tools | Synthetic gRNAs with chemical modifications [72], Ribonucleoprotein (RNP) complexes [74] | Modulate persistence of editing activity; reduce off-target potential | RNP delivery offers transient activity; chemical modifications enhance stability |
| Control Materials | Wild-type and mutant isogenic cell lines [74], Fluorescently labeled tracrRNA [74] | Control for transfection efficiency and establish specificity baseline | Essential for demonstrating allele-specific editing |
The mitigation of CRISPR off-target effects requires a multi-faceted approach combining computational prediction, careful experimental design, and innovative molecular tools. High-fidelity Cas variants like HiFi Cas9 and AI-designed editors such as OpenCRISPR-1 represent significant advancements in achieving the specificity required for therapeutic applications [74] [44]. When combined with optimized gRNA design, appropriate delivery methods, and comprehensive assessment protocols, these tools enable researchers to minimize off-target risks while maintaining therapeutic efficacy. As CRISPR-based therapies continue to advance through clinical trials, rigorous off-target assessment remains paramount for ensuring both scientific validity and patient safety [73] [26]. The field continues to evolve with new detection methods, enhanced computational prediction algorithms, and increasingly sophisticated CRISPR systems that promise to further improve the precision of genome editing.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 system has revolutionized genetic engineering by providing researchers with an unprecedented ability to modify genomes with high precision. However, the efficacy of this technology is not uniform across all genomic loci. The eukaryotic genome is organized into chromatin, a complex structure of DNA and proteins, whose state significantly influences the accessibility and success of CRISPR-mediated editing. This whitepaper addresses two principal structural challenges that impede editing efficiency: GC-rich genomic regions and heterochromatic domains.
GC-rich sequences and tightly packed heterochromatin present substantial barriers that can reduce mutagenesis rates by up to seven-fold [77]. These challenges are particularly relevant for researchers aiming to edit genes involved in regulation, imprinting, and those implicated in various diseases, as these genes are often embedded within such refractory genomic architectures. Understanding the mechanistic basis of these limitations and implementing strategies to overcome them is therefore essential for advancing both basic research and therapeutic applications of CRISPR technology [78] [79].
Mechanistic Challenges to Genome Editing
The GC-Rich Sequence Challenge
GC-rich regions are DNA sequences with a high guanine-cytosine content. These sequences pose multiple distinct challenges for CRISPR-Cas9 editing:
- Secondary Structure Formation: GC-rich single-stranded DNA, including the single-guide RNA (sgRNA) itself, is prone to forming stable secondary structures such as G-quadruplexes and hairpins. These structures can prevent the sgRNA from properly hybridizing with its target DNA sequence, thereby reducing the formation of the Cas9-sgRNA-DNA complex essential for cleavage [79].
- Biochemical Constraints: The Cas9 enzyme requires DNA melting to interrogate the target site. The three hydrogen bonds in GC base pairs, compared to two in AT pairs, confer greater thermodynamic stability to the DNA duplex, making it more energetically costly for Cas9 to unwind and access the target sequence [79].
- Validation Complications: From an experimental perspective, GC-rich regions are notoriously difficult to amplify via PCR and sequence accurately. This "sequence slippage" can lead to mispairing during amplification and poor-quality sequencing data, making it challenging to genotype edited cells and conclusively validate editing outcomes [79].
The Heterochromatin Barrier
In eukaryotic nuclei, DNA is packaged with histone proteins into chromatin. Euchromatin is a relatively open, transcriptionally active form, while heterochromatin is a condensed, transcriptionally repressive form associated with specific histone modifications like H3K9me3 and H4K20me3 [77]. This physical compaction acts as a steric hindrance, limiting the access of the CRISPR-Cas9 machinery to the underlying DNA.
Crucially, the inhibitory effect of heterochromatin is not absolute but kinetic. Research exploiting imprinted genes has demonstrated that heterochromatin can impede mutagenesis by up to 7-fold, but this effect is most pronounced when intracellular Cas9 expression is low and exposure time is brief [77]. Under conditions of prolonged or high-level Cas9 exposure, the mutation frequencies between active and repressed alleles can become comparable, indicating that heterochromatin delays but does not always prevent the endpoint of mutagenesis [77]. Furthermore, the chromatin state does not appear to significantly alter the spectrum of resulting mutations, such as the distribution of insertions and deletions (InDels) or the efficiency of homology-directed repair (HDR) once a double-strand break is successfully introduced [77].
The diagram below illustrates how chromatin structure acts as a kinetic barrier to editing.
Quantitative Analysis of Editing Barriers
The following table summarizes key quantitative findings from research investigating the impact of chromatin state on CRISPR-Cas9 editing efficiency.
Table 1: Quantitative Impact of Heterochromatin on CRISPR-Cas9 Mutagenesis
| Experimental System | Editing Impediment | Key Conditioning Factor | Effect on Repair Outcome | Reference |
|---|---|---|---|---|
| Imprinted genes in mESCs | Up to 7-fold reduction | Low Cas9 concentration & brief exposure | No significant effect on HDR efficiency or InDel spectrum | [77] |
| KvDMR1 locus (sgKVDMR1) | 1.6-fold reduction | High Cas9 concentration & prolonged exposure | Similar endpoint mutation frequency on active and repressed alleles | [77] |
| Correlation with CpG methylation | r = 0.82 (p < 0.05) | Degree of epigenetic repression | Allele-specific mutation bias is lost upon loss of imprinting (LOI) | [77] |
Experimental Strategies and Protocols
To successfully edit difficult-to-access genes, researchers must employ a combination of strategic design, chemical intervention, and robust validation. The following workflow and toolkit outline a comprehensive approach.
A Workflow for Editing Hard-to-Access Genomic Regions
The diagram below outlines a systematic experimental workflow, from initial analysis to final validation, designed to maximize the chances of successfully editing a refractory locus.
The Scientist's Toolkit: Key Reagent Solutions
This table catalogs essential reagents and their specific functions for overcoming the challenges associated with GC-rich and heterochromatic regions.
Table 2: Research Reagent Solutions for Difficult-to-Edit Genes
| Reagent / Tool | Primary Function | Application Context | Key Consideration |
|---|---|---|---|
| Chromatin Modulating Compounds (e.g., HDAC inhibitors like Trichostatin A) | Induces a more open chromatin state by increasing histone acetylation, enhancing Cas9 access [77]. | Pre-treatment of cells prior to and during transfection. | Can have pleiotropic effects on cell physiology; titrate for optimal effect. |
| Cas9-Recruited Chromatin Modulators (e.g., dCas9 fused to KRAB or VP64) | Actively remodels the local chromatin environment; KRAB promotes silencing, while VP64 promotes opening. | Used to pre-condition the target site before introducing the active editing nuclease. | Requires a two-step transfection protocol. |
| High-Fidelity Cas9 Variants (e.g., SpCas9-HF1, eSpCas9(1.1)) | Reduces off-target effects, which is crucial when using high nuclease concentrations to overcome chromatin barriers. | All editing contexts, especially therapeutic applications. | May have slightly reduced on-target activity on some loci. |
| sgRNA Modifications (e.g., chemical modifications on the sgRNA backbone) | Increases sgRNA stability and resistance to secondary structure formation. | Particularly useful for GC-rich targets where sgRNA structure is a problem. | Can be more costly than standard in vitro transcription. |
| Specialized Polymerases (e.g., Q5 High-Fidelity, GC-Rich Enhancers) | Improves amplification efficiency and accuracy of GC-rich templates for genotyping and validation. | Post-editing PCR amplification and sequencing. | Essential for reliable validation of edits in difficult sequences. |
| Bioinformatics Tools (e.g., ICE Analysis, DepMap, primer design tools for GC-rich loci) | Analyzes CRISPR editing efficiency and zygosity from complex sequencing data; assesses gene essentiality. | Experimental design and post-hoc analysis of editing outcomes. | Critical for accurate interpretation of editing success, especially in polyploid systems [79]. |
Detailed Methodologies for Key Experiments
Protocol 1: Assessing Allele-Specific Editing in an Imprinted System (Adapted from [77])
This protocol leverages naturally occurring epigenetic heterogeneity to quantify the effect of chromatin state.
- Cell Culture: Culture mouse embryonic stem cells (mESCs) from F1 hybrid crosses (e.g., C57BL/6J × JF1) in serum-free conditions with GSK3β inhibitors to maintain imprinting stability.
- Epigenetic Status Validation:
- Perform allele-specific chromatin immunoprecipitation (ChIP) for heterochromatin marks (H3K9me3, H4K20me3) on the target locus.
- Perform allele-specific DNase-I hypersensitivity assays.
- Quantify CpG methylation at the target site via bisulfite sequencing in mock-transfected cells to establish the baseline epigenetic state.
- Transfection and Selection: Transfect cells with a plasmid expressing Cas9 and the target sgRNA (e.g., pX459v2). Include a single-stranded oligodeoxynucleotide (ssODN) donor template containing a blocking mutation to prevent re-cleavage. Select transfected cells with puromycin for 96 hours.
- Amplicon Sequencing and Analysis: Harvest cells as a pool. Amplify the target region by PCR and subject to Illumina sequencing. Use strain-specific SNPs within the amplicon to bioinformatically separate and quantify the frequency and spectrum of mutations on the maternal (typically repressed) and paternal (typically active) alleles.
Protocol 2: Overcoming GC-Rich and Heterochromatic Barriers in Cell Lines
This is a generalized protocol for editing a difficult endogenous locus in mammalian cell lines.
- sgRNA Design and Validation:
- Use multiple sgRNAs targeting different regions within the same gene. Prioritize protospacers with intermediate (40-60%) GC content to avoid stability and specificity issues.
- In silico screen for potential off-target sites.
- Clonally produce sgRNAs via in vitro transcription or purchase synthetically modified sgRNAs for enhanced stability.
- Chromatin Pre-Conditioning (Optional but Recommended):
- Treat cells with a chromatin-modifying agent 24 hours before transfection. For example, use 100 nM Trichostatin A (an HDAC inhibitor) to promote chromatin opening.
- Alternatively, for a more targeted approach, pre-transfect with a plasmid encoding dCas9 fused to a transcriptional activator (like VP64) and a sgRNA targeting the immediate vicinity of the desired cut site. Allow 48-72 hours for chromatin remodeling.
- Delivery of Editing Machinery:
- For plasmid-based delivery, use a high-concentration transfection protocol and extend the time between transfection and analysis to 96-120 hours to allow more time for the nuclease to access the target.
- For ribonucleoprotein (RNP) delivery, complex 2-4 µg of purified Cas9 protein with a 1:2 molar ratio of sgRNA. Deliver via nucleofection for high efficiency and rapid activity, which can help mitigate the kinetic barrier of heterochromatin.
- Enhanced Genotyping:
- Extract genomic DNA and set up PCRs with a polymerase specifically engineered for high GC content, using a manufacturer-supplied GC-rich buffer.
- Use a touchdown PCR program to improve specificity.
- For sequencing, consider cloning PCR products or utilizing long-read sequencing technologies (e.g., PacBio) to accurately resolve complex edits and avoid artifacts from repetitive or GC-skewed regions.
The challenges posed by GC-rich sequences and heterochromatin are significant but surmountable barriers in CRISPR research. The key insight is that heterochromatin acts primarily as a kinetic barrier that can be overcome by optimizing delivery and exposure, while GC-richness requires careful molecular handling and validation. By integrating the strategies outlined herein—including informed sgRNA design, strategic chromatin modulation, robust delivery methods, and meticulous validation—researchers can significantly enhance their ability to edit previously intractable genomic loci. This expands the frontiers of CRISPR application, enabling more comprehensive functional genomics and accelerating the development of advanced genetic therapies.
The advent of CRISPR-Cas9-based genome editing revolutionized genetics by providing a precise method for altering DNA sequences, offering a convenient alternative to complex techniques like zinc-finger nucleases [80]. However, when investigating essential genes—those critical for cellular survival—traditional CRISPR knockout (CRISPRko) approaches face significant limitations. Complete disruption of essential gene function often leads to cellular toxicity or lethality, precluding functional analysis [80]. This fundamental constraint has driven the development of complementary techniques that modulate gene expression without permanent DNA damage, primarily CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) [81].
These technologies represent a paradigm shift from the binary "on-off" switch of gene knockout to a more nuanced "dimmer switch" approach that allows fine-tuning of gene expression levels [80]. This capability is particularly valuable for studying essential genes, modeling the partial reduction of gene expression often achieved by pharmacological interventions, and understanding dose-dependent gene functions [80] [82]. The flexibility of CRISPRi and CRISPRa has established them as powerful tools for functional genomics, both in basic research and drug development contexts [83] [81].
Fundamental Mechanisms: From DNA Cutting to Transcriptional Control
The dCas9 Foundation
CRISPRi and CRISPRa systems fundamentally differ from traditional CRISPR-Cas9 genome editing through their use of a catalytically dead Cas9 (dCas9). This engineered variant contains point mutations (typically D10A and H840A) that inactivate the RuvC and HNH nuclease domains, rendering the protein incapable of cleaving DNA [80] [84]. However, dCas9 retains its ability to bind specific DNA sequences guided by a single-guide RNA (sgRNA), serving as a programmable platform for recruiting effector domains to precise genomic locations [83].
CRISPR Interference (CRISPRi) Mechanism
CRISPRi functions as a transcriptional repressor system. When targeted to promoter regions, dCas9 alone can achieve modest repression (60-80%) in mammalian cells by sterically hindering RNA polymerase binding or transcriptional elongation [80]. Significantly enhanced repression is achieved by fusing dCas9 to repressor domains such as the Krüppel-associated box (KRAB) domain from KOX1 [83] [84]. KRAB recruits additional chromatin-modifying factors that establish transcriptionally repressive heterochromatin, effectively silencing target gene expression in a reversible, titratable, and non-toxic manner [80] [83].
CRISPR Activation (CRISPRa) Mechanism
Conversely, CRISPRa harnesses dCas9 to activate transcription. Early implementations used simple fusions to activator domains like VP64, but these achieved only modest activation [83]. More potent systems have been developed using three principal strategies:
- Direct fusions to dCas9, exemplified by the VPR approach (a tripartite fusion of VP64, p65, and Rta activation domains) [83].
- Protein scaffolds, such as the SunTag system, which uses a tandem array of peptide epitopes to recruit multiple activator domains [80] [83].
- RNA scaffolds, exemplified by the Synergistic Activation Mediator (SAM) system, where engineered RNA aptamers in the sgRNA recruit MS2 coat protein-fused activators (p65 and HSF1) [83] [85].
Table 1: Comparison of Key CRISPR Technologies
| Feature | CRISPR Knockout (CRISPRko) | CRISPR Interference (CRISPRi) | CRISPR Activation (CRISPRa) |
|---|---|---|---|
| Cas9 Form | Wild-type (nuclease active) | dCas9 fused to repressors (e.g., KRAB) | dCas9 fused to activators (e.g., VP64, SAM) |
| Mechanism of Action | Creates double-strand breaks, introduces indels | Blocks transcription, induces heterochromatin | Recruits transcriptional machinery |
| Genetic Change | Permanent mutation | Reversible expression modulation | Reversible expression modulation |
| Effect on Expression | Complete loss-of-function | Tunable knockdown (up to 80-99% repression) | Tunable overexpression (up to 1000-fold activation) |
| Best For | Complete gene disruption, non-essential genes | Essential genes, partial knockdown, non-coding RNAs | Gain-of-function, gene dosage studies, non-coding RNAs |
Experimental Design and Workflow
Guide RNA Design Considerations
A critical distinction between CRISPRko and CRISPRi/a experiments lies in sgRNA design. For CRISPRko, efficient sgRNAs typically target early exons to maximize frameshift potential. In contrast, CRISPRi and CRISPRa sgRNAs must target specific regions relative to the transcriptional start site (TSS) [84].
Systematic screens have identified optimal targeting windows for both technologies [84]:
- CRISPRi: gRNAs within -50 to +300 base pairs from the TSS, with maximal efficacy in the first 100 bp downstream of the TSS.
- CRISPRa: gRNAs within -400 to -50 bp upstream of the TSS.
Additional factors affecting sgRNA efficacy include protospacer length (avoid >21 bp), avoidance of nucleotide homopolymers, and local chromatin accessibility [84]. For genome-scale screens, libraries typically include 3-10 sgRNAs per gene to ensure robust phenotype detection despite variations in individual sgRNA activity [83] [84].
Stable Cell Line Generation
For consistent and robust perturbations, especially in large-scale screens, generating stable "helper" cell lines that express the dCas9-effector fusion is recommended [84]. Recent advancements have significantly improved the efficiency of this process:
- Self-selecting CRISPRa systems (CRISPRa-sel) combine piggyBac transposon technology with a CRISPRa-dependent puromycin resistance gene, enabling rapid production of uniform, potent CRISPRa-competent cell populations without laborious single-cell cloning [85].
- Inducible systems provide temporal control over CRISPRi/a activity. Novel drug-responsive systems (e.g., iCRISPRa/i) fuse mutated human estrogen receptor (ERT2) domains to CRISPRa/i components, enabling rapid, reversible nuclear translocation of the effectors upon 4-hydroxytamoxifen (4OHT) treatment [86]. These systems offer lower baseline leakage and faster response compared to earlier tetracycline-inducible systems [86].
Research Reagent Solutions
Table 2: Essential Research Reagents for CRISPRi/a Experiments
| Reagent Category | Specific Examples | Function & Importance |
|---|---|---|
| dCas9 Effector Systems | dCas9-KRAB (for CRISPRi), SAM system (for CRISPRa), dCas9-VPR | Core machinery for gene repression or activation; system choice impacts efficiency and dynamic range [83] [85] [84]. |
| Delivery Vectors | Lentiviral vectors, piggyBac transposon systems | Enable stable integration of large CRISPRa/i cassettes into host cell genome; crucial for persistent expression [85] [84]. |
| sgRNA Libraries | Genome-scale pooled libraries (e.g., Dolcetto for CRISPRi, Calabrese for CRISPRa) | Collections of sgRNAs targeting entire genomes; designed with optimal positioning relative to TSS [83] [86]. |
| Inducible Systems | iCRISPRa/i (4OHT-responsive), Tet-based systems (doxycycline-responsive) | Provide temporal control over gene perturbation; essential for studying essential genes and dynamic processes [86]. |
| Selection Markers | Puromycin resistance, fluorescent reporters (GFP, mCherry) | Enable enrichment of successfully transduced cells; critical for generating uniform, high-efficiency cell populations [85] [86]. |
Applications in Functional Genomics and Drug Discovery
Genetic Screens for Essential Genes
CRISPRi and CRISPRa excel in functional genomic screens to identify genes involved in specific biological processes or disease states [83]. Pooled screens using genome-scale sgRNA libraries have been particularly powerful for investigating essential genes:
- Fitness/viability screens identify genes essential for cell growth and survival under baseline conditions or specific contexts [83]. For example, CRISPRi screens in human induced pluripotent stem cells (hiPSCs) have revealed distinct dependencies on mRNA translation machinery and quality control pathways compared to differentiated cells [87].
- Chemical-genetic interactions/sensitization screens identify genes whose perturbation modifies cellular response to drugs, toxins, or other selective pressures [83]. These screens can reveal drug mechanism of action, resistance mechanisms, and synergistic targets for combination therapies [83] [82].
A key advantage of CRISPRi in essential gene studies is the ability to achieve partial knockdowns that reduce but do not eliminate gene function, allowing cell survival and revealing phenotypes that would be masked by complete knockout [80]. CRISPRa screens can identify genes whose overexpression confers selective advantages or disadvantages, often revealing tumor suppressors and developmental regulators [83].
Modeling Disease and Development
The reversible, titratable nature of CRISPRi/a makes them ideal for modeling dynamic biological processes:
- Studying essential genes in development: CRISPRi enables knockdown of developmentally critical genes without embryonic lethality, facilitating analysis of their roles in later stages [86].
- Cell-type-specific essentiality: Comparative CRISPRi screens across hiPSCs and differentiated lineages (neurons, cardiomyocytes) have revealed how essentiality of core cellular machinery components varies with cell identity [87].
- Endogenous gene activation: CRISPRa enables physiological overexpression of genes in their native genomic context, including correct splice variants, which is particularly valuable for large transcripts and non-coding RNAs difficult to study with cDNA overexpression [80] [84].
Therapeutic Applications and Target Validation
CRISPRi and CRISPRa hold significant promise for therapeutic development:
- Target identification and validation: CRISPRi/a screens can identify high-value therapeutic targets by linking gene expression changes to disease-relevant phenotypes [82] [81].
- Disease modeling: CRISPRa/i can create more accurate cellular models of genetic disorders by modulating expression of disease-associated genes without permanent genomic alteration [81].
- Therapeutic modality: Direct therapeutic application of CRISPRi/a is being explored for diseases where transcriptional modulation could provide benefit, with preclinical successes in models of epilepsy, muscular dystrophy, retinitis pigmentosa, and cancer [88] [81].
A significant therapeutic advantage of CRISPRa is its independence from specific disease-causing mutations, potentially making it applicable to broader patient populations compared to mutation-correcting approaches [88].
Comparative Analysis with Alternative Technologies
CRISPRi vs. RNA Interference (RNAi)
While both technologies achieve gene knockdown, they differ fundamentally:
- Mechanism: RNAi operates post-transcriptionally by degrading cytoplasmic mRNA, while CRISPRi acts at the transcriptional level in the nucleus [84].
- Specificity: CRISPRi generally exhibits fewer sequence-specific off-target effects than RNAi [80] [84].
- Scope: CRISPRi can effectively target both protein-coding and non-coding genes, while RNAi struggles with nuclear non-coding RNAs [80] [84].
- Efficacy: CRISPRi typically generates more robust and reproducible phenotypes in large-scale screens [84].
CRISPRa vs. ORF Overexpression
Traditional gain-of-function approaches using open reading frame (ORF) expression vectors have limitations that CRISPRa addresses:
- Expression levels: ORF overexpression often uses strong viral promoters leading to supraphysiological expression, while CRISPRa activates genes from endogenous promoters at more physiological levels [84].
- Splice variants: CRISPRa naturally upregulates all endogenous splice variants, while ORF overexpression typically expresses only a single cDNA-defined isoform [84].
- Library generation: Genome-scale CRISPRa libraries are more straightforward to synthesize and handle than ORF libraries [84].
Technical Challenges and Optimization Strategies
Despite their powerful applications, CRISPRi and CRISPRa implementations face several technical challenges with corresponding optimization approaches:
- Guide RNA efficacy: Variable activity among sgRNAs remains a significant challenge. Solution: Use validated sgRNA libraries with multiple guides per gene, and employ optimized sgRNA scaffolds that enhance functionality, such as modified SAM-compatible scaffolds that improve activation efficiency [85].
- Chromatin accessibility: Dense chromatin structure can limit dCas9 binding to target sites. Solution: Combine multiple sgRNAs targeting the same gene or use chromatin-modifying domains in the activator/repressor systems [80] [84].
- Delivery limitations: The large size of CRISPRa/i cassettes challenges viral delivery, particularly with adeno-associated viruses (AAVs) which have limited cargo capacity [88]. Solution: Use split systems, smaller Cas9 orthologs, or non-viral delivery methods such as piggyBac transposons [85].
- Off-target effects: Although generally more specific than RNAi, CRISPRi/a can still have off-target effects. Solution: Use carefully designed sgRNAs with computational off-target prediction, and employ inducible systems to limit exposure time [86].
CRISPRi and CRISPRa have fundamentally expanded our toolbox for genetic manipulation, providing powerful alternatives to traditional knockout approaches, particularly for studying essential genes. By enabling reversible, titratable control of gene expression without permanent genomic alteration, these technologies offer more physiologically relevant models of gene dosage effects and better mimic partial inhibition achieved by pharmacological interventions [80] [82].
The continued refinement of CRISPRi/a systems—through improved effector domains, optimized delivery strategies, and enhanced temporal control—promises to further accelerate their adoption in both basic research and therapeutic development [81] [86]. As these technologies mature, they will undoubtedly yield deeper insights into gene function, disease mechanisms, and novel therapeutic opportunities across the spectrum of human disorders.
For researchers navigating the complexities of essential gene function, CRISPRi and CRISPRa represent not just alternatives to knockout, but essential complementary approaches that provide a more complete understanding of gene function in health and disease.
Optimizing for Ploidy and Copy Number Variation in Complex Cell Lines
The foundational principle of CRISPR gene editing research is to establish a precise genotype-to-phenotype relationship by introducing targeted genetic perturbations. However, this principle encounters significant challenges when applied to complex cell lines with variable ploidy and copy number variations (CNVs). Ploidy refers to the number of complete sets of chromosomes in a cell, ranging from haploid (single copy) to polyploid (many copies), while CNVs represent regions of the genome where the number of copies of a particular DNA segment varies from one individual to another [79]. In humans, approximately 12% of the genome contains CNVs, with each individual typically harboring about 12 CNVs [79].
The efficiency of CRISPR editing is profoundly influenced by these genomic complexities. In simple diploid systems, researchers need to edit only two alleles to achieve complete gene knockout, but in polyploid systems or regions with CNVs, the same gene may be present in multiple copies, making complete genetic alteration substantially more challenging [79]. This technical challenge is particularly relevant in cancer research, where many cell lines exhibit extensive aneuploidy and CNVs. Understanding and optimizing for these complexities is therefore essential for rigorous CRISPR experimental design and interpretation, particularly in disease modeling and drug discovery applications [89].
Fundamental Principles: How Ploidy and CNVs Impact CRISPR Outcomes
Mechanistic Challenges in Complex Genomes
The presence of multiple gene copies creates several interconnected challenges for CRISPR editing. First, the sheer number of target alleles requires highly efficient editing to achieve complete knockout. Second, heterogeneity in editing outcomes across different copies can complicate phenotypic analysis, as unedited wild-type copies may compensate for edited ones [79]. Third, and perhaps most importantly, research has revealed that CRISPR-Cas9 targeting within amplified genomic regions can elicit a gene-independent anti-proliferative cell response [89].
This phenomenon was systematically demonstrated in a genome-scale loss-of-function screen across 33 cancer cell lines, where targeting genes within amplified regions—including both expressed and unexpressed genes—led to significantly decreased cell proliferation through induction of a G2 cell cycle arrest [89]. The response correlated strongly with the number of target loci rather than the specific gene function, confounding the interpretation of essentiality screens in amplified regions. This effect was not observed in parallel RNAi screens, suggesting it is specific to DNA-breaking mechanisms rather than transcript degradation [89].
Table 1: Impact of Genomic Complexity on CRISPR Editing Efficiency
| Genomic Feature | Effect on CRISPR Editing | Experimental Consequence |
|---|---|---|
| High Ploidy (e.g., tetraploid, polyploid) | Multiple alleles require editing; reduced likelihood of complete knockout | Residual wild-type function; potential false negatives in knockout screens |
| Copy Number Variations (CNVs) | Variable copy number between cell lines; requirement to edit all amplified copies | Inconsistent editing efficiency across models; confounded essentiality scores |
| Segment Duplications | Homologous sequences increase off-target risk | Unintended editing at paralogous loci; complex rearrangement patterns |
Quantitative Relationship Between Copy Number and Editing Outcomes
The relationship between copy number and CRISPR effectiveness follows a predictable pattern. Cells with higher copy numbers of a target gene demonstrate increased sensitivity to CRISPR-induced DNA damage, but this effect is independent of the gene's function and rather depends on the cumulative number of double-strand breaks [89]. This creates a particular challenge in cancer cell lines where oncogenes are frequently amplified—the apparent "essentiality" of these genes in CRISPR screens may reflect this general DNA damage response rather than true biological essentiality.
Table 2: Comparison of Genetic Perturbation Methods in Amplified Genomic Regions
| Perturbation Method | Mechanism of Action | Performance in Amplified Regions | Key Limitations |
|---|---|---|---|
| CRISPR Knockout | Creates double-strand breaks via Cas9 nuclease | Induces gene-independent anti-proliferative response; confounds essentiality interpretation [89] | Cellular response to multiple DNA breaks; difficult multi-copy editing |
| CRISPR Interference | dCas9 fusion proteins block transcription | More accurate essentiality assessment; no DNA damage [79] | Transient suppression; potential incomplete repression |
| RNA Interference | Degrades mRNA via RNAi machinery | No DNA damage response; identifies specific drivers in amplifications [89] | Off-target transcriptional effects; incomplete knockdown |
Methodological Framework: Experimental Strategies for Optimization
Pre-Experimental Characterization of Model Systems
Successful CRISPR editing in complex cell lines begins with comprehensive genomic characterization. The following workflow provides a systematic approach to cell line validation and editing optimization:
Karyotyping and Ploidy Analysis
Begin with standard karyotyping to identify chromosomal abnormalities in quantity and structure. Many commonly used cell lines, including 293 (HEK-293) and hTERT RPE-1, are not perfect diploids but rather "hypotriploid" or "near-diploid," having more than two sets of chromosomes on average [79]. Understanding the baseline ploidy is essential for designing appropriate editing strategies and interpreting results.
CNV Profiling
Utilize real-time quantitative PCR (qPCR) as a relatively inexpensive and fast method to quantify copy number variations for your genes of interest [79]. For broader profiling, whole-genome sequencing or SNP arrays can identify CNV landscapes across the entire genome. The Broad Institute-Novartis Cancer Cell Line Encyclopedia (CCLE) provides ABSOLUTE DNA copy number data for many cancer cell lines, which can serve as a valuable resource [89].
Essential Gene Assessment
Consult the Dependency Map (DepMap) portal to determine if your target gene is classified as "common essential" [79]. If it is, consider alternative approaches such as CRISPR interference (CRISPRi) or RNAi-based knockdown instead of complete knockout, or generate heterozygous knockouts that retain one functional copy to maintain cell viability [79].
Advanced Editing Strategies for Complex Genomic Loci
Multiplexed Editing Approaches
For polyploid cell lines or genes with high copy number, simultaneous targeting with multiple guide RNAs can increase the probability of complete knockout. Advances in multiplexed CRISPR systems enable the delivery of several gRNAs targeting the same gene or multiple genes simultaneously [90]. The CDKO (CRISPR-based double-knockout) library system utilizes dual gRNAs expressed from different promoters (e.g., human U6 and mouse U6) to avoid recombination between identical sequences [90].
For particularly challenging repetitive regions, the SCORE (single-guide CRISPR/Cas targeting of repetitive elements) system has been developed to model reciprocal CNVs by targeting segmental duplications, successfully generating reciprocal CNVs at chromosome 16p11.2 and 15q13.3 regions associated with neurodevelopmental disorders [91].
Alternative CRISPR Modalities
When complete knockout is problematic in polyploid systems, consider these alternative approaches:
- CRISPR Interference: Using nuclease-dead Cas9 fused to repressor domains to reduce gene expression without permanent DNA alteration [79]
- Base Editing: Making precise single-nucleotide changes without double-strand breaks, potentially creating hypomorphic alleles
- CRISPR Nickases: Using Cas9 variants that cut only one DNA strand to reduce off-target effects while maintaining on-target efficiency [90]
Practical Implementation: Reagents and Validation Methods
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Optimizing CRISPR in Complex Cell Lines
| Reagent / Tool | Function | Application Notes |
|---|---|---|
| Karyotyping Kits | Determines chromosome number and structure | Establish baseline ploidy before editing experiments |
| qPCR Assays | Quantifies gene copy number variations | Fast, cost-effective CNV validation for specific loci |
| GeCKOv2 Library | Genome-scale CRISPR knockout screening | Identifies essential genes; contains 123,411 sgRNAs targeting 19,050 genes [89] |
| CDKO Library | Paired gRNA library for large deletions | Uses human U6 and mouse U6 promoters to avoid recombination [90] |
| ICE Bioinformatics Tool | Analyzes CRISPR editing efficiency | Determines zygosity in edited clones; critical for polyploid lines [79] |
| dCas9-KRAB Constructs | CRISPR interference for gene suppression | Enables knockdown without DNA damage; ideal for essential genes [79] |
| Lipid Nanoparticles | Delivery of CRISPR components | Particularly efficient for liver targets; enables redosing [26] |
Validation and Analysis Methods
Editing Efficiency Assessment
The Inference of CRISPR Edits (ICE) tool from Synthego provides critical analytical capabilities for determining editing efficiency and zygosity in complex cell lines [79]. This bioinformatics platform helps deconvolve mixed editing outcomes in polyploid systems, where multiple alleles may show different indels.
Functional Validation
For essential genes in amplified regions, where complete knockout is lethal, consider these functional validation approaches:
- Heterozygous Knockouts: Maintain cell viability while reducing gene dosage
- Conditional Knockouts: Use inducible systems or site-specific recombinases
- Complementation Assays: Reintroduce wild-type or mutant cDNA to confirm phenotype specificity
The following diagram illustrates the cellular response mechanisms to CRISPR editing in amplified genomic regions:
Emerging Solutions and Future Directions
Recent technological advances are providing new solutions to the challenges of editing complex genomic regions. The development of bridge recombinase systems like ISCro4 enables programmable human genome editing through targeted insertions, inversions, and excisions without relying on the endogenous DNA repair machinery that complicates editing in polyploid systems [92]. This system has achieved 20% insertion efficiency and 82% genome-wide specificity, successfully moving DNA segments up to nearly one megabase in size [92].
For therapeutic applications in highly polyploid tissues, new delivery systems are showing promise. Lipid nanoparticles (LNPs) have proven particularly effective for liver-directed therapies, as they naturally accumulate in hepatic tissue and can be redosed without the immune reactions associated with viral vectors [26]. In clinical trials for hereditary transthyretin amyloidosis (hATTR), LNP-delivered CRISPR therapies have achieved sustained 90-92% reductions in disease-causing protein levels over 24 months [26].
In cancer research, where ploidy and CNV complexity are greatest, novel screening approaches are being developed. The VECOS (virus-encoded CRISPR screening) system encodes sgRNA libraries directly in viral genomes, allowing direct measurement of gene perturbation effects on viral propagation rather than relying solely on cell survival as a readout [92]. This provides more nuanced insights into gene function in complex genomic contexts.
As CRISPR technology continues to evolve, the integration of more sophisticated computational tools and single-cell analytical methods will further enhance our ability to navigate the complexities of ploidy and copy number variation, ultimately strengthening the fundamental principle of establishing clear genotype-phenotype relationships through precise genetic editing.
CRISPR-Cas9 genome editing has revolutionized biological research and therapeutic development by enabling precise genetic modifications. However, a significant challenge persists: the potential for off-target effects where unintended genomic locations are cleaved. These off-target cleavage events can lead to deleterious consequences such as loss of gene function, chromosomal rearrangements, and potentially oncogenic transformations, presenting substantial safety concerns for clinical applications [93] [94]. The development of robust, unbiased methods to identify these off-target sites is therefore paramount for advancing CRISPR-based therapeutics.
This technical guide examines two pivotal genome-wide approaches for off-target profiling: GUIDE-Seq (Genome-wide Unbiased Identification of DSBs Enabled by Sequencing), an in cellulo method, and CIRCLE-Seq (Circularization for In Vitro Reporting of CLeavage Effects by Sequencing), an in vitro method. These techniques have become cornerstone technologies in the field, providing comprehensive specificity landscapes for CRISPR nucleases that enable researchers to select optimal guide RNAs and assess the safety profile of their editing systems prior to clinical use [93] [95].
Methodological Principles and Workflows
GUIDE-Seq: In Vivo Double-Strand Break Tagging
GUIDE-Seq operates on the principle of capturing double-stranded breaks (DSBs) within living cells through the integration of a specialized double-stranded oligodeoxynucleotide (dsODN) tag. The method comprises two distinct stages:
Stage I: DSB Tagging in Living Cells During active genome editing, blunt-ended DSBs generated by CRISPR-Cas9 are tagged via integration of a 34 bp blunt, 5' phosphorylated dsODN through the non-homologous end joining (NHEJ) repair pathway. Critical to the method's efficiency is the incorporation of phosphorothioate linkages at both the 5' and 3' ends of the dsODN, which significantly stabilizes the oligonucleotide against cellular degradation and enables robust integration efficiencies approaching those of native indel formation [93].
Stage II: Tagged Site Amplification and Sequencing Genomic DNA is extracted and randomly sheared, followed by ligation of single-tailed next-generation sequencing adapters. The core innovation of GUIDE-Seq lies in the Single-Tail Adapter/Tag (STAT)-PCR method, which uses one primer specific to the integrated dsODN and another that anneals to the sequencing adapter. This approach enables specific unidirectional amplification of sequences adjacent to dsODN integration sites while minimizing background amplification. Incorporation of a random 8 bp molecular barcode during amplification facilitates PCR bias correction and accurate quantitation of unique sequencing reads [93].
Table 1: Key Research Reagents for GUIDE-Seq
| Research Reagent | Function | Technical Specifications |
|---|---|---|
| Double-stranded ODN (dsODN) | DSB tagging via NHEJ | 34 bp, blunt-ended, 5' phosphorylated, phosphorothioate linkages at 5' and 3' ends |
| STAT-PCR primers | Amplification of tagged sites | Includes dsODN-specific primer and adapter-specific primer |
| Molecular barcodes | PCR bias correction | Random 8 bp sequences |
| Sequencing adapters | Library preparation | Single-tailed design for unidirectional amplification |
CIRCLE-Seq: In Vitro Cleavage Detection
CIRCLE-Seq employs a fundamentally different approach that occurs entirely in vitro, eliminating the need for cellular delivery of editing components. Its workflow centers on strategic circularization of genomic DNA to dramatically reduce background noise:
Genomic DNA Preparation and Circularization High molecular weight genomic DNA is extracted from cells of interest and fragmented by sonication. The resulting fragments are subsequently circularized using circligase, creating covalently closed DNA molecules. This circularization step is crucial as it physically separates Cas9-cleavable sites (within the circles) from the vast majority of non-cleavable genomic DNA [95].
In Vitro Cleavage and Library Preparation The circularized DNA library is incubated with preassembled Cas9-guide RNA ribonucleoprotein (RNP) complexes under optimized cleavage conditions. Active Cas9 cleaves only those circularized molecules containing complementary target sequences, linearizing them in the process. The newly generated ends of these linearized molecules are then ligated with sequencing adapters and prepared for high-throughput sequencing. Since only Cas9-cleaved fragments receive adapters, the background is dramatically reduced compared to other in vitro methods [96] [95].
Table 2: Key Research Reagents for CIRCLE-Seq
| Research Reagent | Function | Technical Specifications |
|---|---|---|
| Circligase enzyme | DNA circularization | Covalently closes linear DNA fragments |
| Cas9 nuclease | In vitro cleavage | Purified protein complexed with guide RNA |
| Guide RNA | Target specificity | In vitro transcribed or synthesized |
| Fragmented genomic DNA | Substrate for cleavage | Sonicated or enzymatically fragmented |
| Sequencing adapters | Library preparation | Compatible with Illumina platforms |
Comparative Performance Analysis
Sensitivity and Specificity Profiles
When directly compared, GUIDE-Seq and CIRCLE-Seq demonstrate complementary strengths and limitations. CIRCLE-seq exhibits exceptional sensitivity in detecting rare off-target events, identifying numerous bona fide off-target sites that occur in human cells but were missed by GUIDE-seq and other cell-based methods [95]. This heightened sensitivity stems from the ability to use high concentrations of Cas9-gRNA complexes in vitro, enabling detection of cleavage sites that might be infrequent in cellular environments.
GUIDE-Seq's primary advantage lies in its biological relevance, as it captures editing outcomes within the native cellular context, including the influence of chromatin structure, DNA repair mechanisms, and cellular physiology. Studies validating GUIDE-Seq have demonstrated that over 80% of identified sites show characteristic NHEJ-mediated indels, confirming their biological relevance [93].
Table 3: Method Comparison Guide
| Parameter | GUIDE-Seq | CIRCLE-Seq |
|---|---|---|
| Cellular context | In cellulo (preserves biological complexity) | In vitro (bypasses cellular barriers) |
| Detection sensitivity | Moderate (can miss low-frequency events) | High (identifies rare cleavage sites) |
| Background signal | Moderate | Very low (180,000-fold enrichment over Digenome-seq) |
| Throughput | Lower (requires cell culture and transfection) | Higher (amenable to screening multiple gRNAs) |
| Relevance to living systems | Direct assessment of cellular editing | Predictive (requires validation in cells) |
| Sequencing depth required | Moderate | Low (works efficiently with benchtop sequencers) |
| Personalization potential | Limited to transfertable cell types | High (can use DNA from any source, including patient samples) |
Technical Considerations for Implementation
GUIDE-Seq Optimization Requirements: Successful GUIDE-Seq implementation requires careful optimization of dsODN delivery and integration. The original protocol achieved robust integration efficiencies only after incorporating phosphorothioate linkages at both the 5' and 3' ends of both dsODN strands. Additionally, transfection efficiency critically impacts success, as poor delivery results in insufficient tag integration for detection [93].
CIRCLE-Seq Advantages in Screening: CIRCLE-seq offers distinct practical advantages for large-scale screening applications. The method requires approximately 100-fold fewer sequencing reads than other in vitro methods like Digenome-seq, making it compatible with benchtop sequencers. Furthermore, CIRCLE-seq does not require a reference genome for initial site identification, enabling off-target profiling in genetically diverse samples or non-model organisms [95].
Recent Advancements and Future Directions
The field of off-target detection continues to evolve with several methodological improvements emerging. GUIDE-tag represents an enhancement of GUIDE-Seq that incorporates tethering between Cas9 nuclease and DNA donor through SpyCas9-mSA and biotin-dsDNA interactions, significantly increasing capture efficiency in mouse liver and lung tissues. This advancement enables detection of off-target sites with editing rates as low as 0.2% in vivo [97].
TEG-Seq (Tag-Enriched GUIDE-Seq) addresses sensitivity limitations in original GUIDE-Seq by using 5' phosphorylated primers for PCR amplification and differential marking of amplicons containing the dsODN tag. This modification reduces nonspecific amplification and improves DSB detection sensitivity, with studies demonstrating detection of 252 off-target events for the HEK4 site compared to 132 with standard GUIDE-Seq [98].
AID-seq represents another recent innovation enabling massively parallel CRISPR off-target detection. This method demonstrates high sensitivity and precision while offering the unique capability for high-throughput sgRNA screening and off-target prediction model building [99].
Concurrently, the development of AI-designed editors like OpenCRISPR-1—generated through large language models trained on CRISPR sequences—showcases how computational approaches are creating editors with improved specificity profiles from the outset [44].
GUIDE-Seq and CIRCLE-Seq have established themselves as cornerstone technologies for comprehensive off-target profiling in CRISPR genome editing. GUIDE-Seq provides biologically relevant specificity landscapes within native cellular environments, while CIRCLE-Seq offers exceptional sensitivity for detecting rare cleavage events in a controlled in vitro setting. The continued refinement of these methods, coupled with emerging computational design approaches, promises to further enhance the safety and precision of CRISPR-based therapeutics, ultimately accelerating their translation to clinical applications.
Researchers should select between these methods based on their specific experimental needs: GUIDE-Seq for assessing editing outcomes in relevant cellular contexts, and CIRCLE-Seq for maximal sensitivity and scalability in off-target site identification. For the most rigorous safety assessment, employing a combination of both approaches may provide complementary insights that neither method could deliver alone.
CRISPR in Context: Validation Techniques and Comparison to ZFNs & TALENs
In CRISPR gene editing, the introduction of double-strand breaks and subsequent repair by cellular mechanisms can lead to a complex mixture of insertion and deletion mutations (indels) and other genomic alterations at the target site. Genotyping validation—the process of precisely characterizing the resulting genetic modifications—is therefore not merely a final confirmatory step but a fundamental component of the research workflow. Without accurate validation, researchers cannot confirm whether intended edits have occurred, assess editing efficiency, or detect potential unintended consequences. The choice of validation method directly impacts the reliability, depth, and scope of experimental conclusions. This technical guide examines the two predominant methodologies for genotyping validation: the established Sanger sequencing and the increasingly powerful Next-Generation Sequencing (NGS), framing their application within the basic principles of CRISPR research.
The foundational principle underlying the need for rigorous validation is the inherent heterogeneity of CRISPR editing outcomes. When the CRISPR-Cas9 complex, guided by a single guide RNA (sgRNA), creates a double-strand break at the target locus, the cell employs primarily the error-prone non-homologous end joining (NHEJ) pathway for repair. This process results in a diverse spectrum of indels at the target site [100]. Consequently, the edited cell population is rarely uniform but is instead a mosaic of different alleles. Accurately quantifying this mixture and identifying the specific mutations present is crucial for interpreting experimental results, whether the goal is functional gene knockout, precise nucleotide alteration, or therapeutic development.
Foundational Technologies: A Comparative Analysis
Sanger Sequencing: The Gold Standard for Targeted Analysis
Sanger sequencing, also known as chain-termination or dideoxy sequencing, operates on a principle of selective chain termination during DNA synthesis. The process involves a DNA polymerase synthesizing a complementary strand from a single-stranded template in the presence of both standard deoxynucleotides (dNTPs) and a low concentration of fluorescently-labeled dideoxynucleotides (ddNTPs). These ddNTPs lack a 3'-hydroxyl group, causing random termination of the growing DNA strand upon their incorporation. The resulting DNA fragments of varying lengths are then separated by capillary gel electrophoresis, and the fluorescent tag on the terminal ddNTP is detected to reveal the DNA sequence [101] [102].
In the context of CRISPR validation, the primary strength of Sanger sequencing lies in its ability to generate long, high-quality contiguous reads—typically between 500 to 1000 base pairs—from a specific PCR amplicon spanning the target site [103] [102]. This allows for direct visualization of the editing locus. However, a significant limitation is its low sensitivity for detecting mixed sequences. The sequencing chromatogram becomes increasingly difficult to interpret clearly when multiple alleles are present, as overlapping fluorescence peaks emerge downstream of the editing site. Its effective limit of detection for a minor allele is typically in the range of 15-20% allele frequency, making it unsuitable for accurately quantifying editing efficiency in a heterogeneous sample or for detecting rare editing events [104].
Next-Generation Sequencing: The Power of Parallelism
Next-Generation Sequencing (NGS), or massively parallel sequencing, represents a paradigm shift from Sanger's linear approach. NGS technologies, such as Illumina's Sequencing by Synthesis (SBS), simultaneously sequence millions to billions of DNA fragments in a single run [105] [104]. The general workflow involves fragmenting DNA, attaching fragments to a solid surface or bead, amplifying them locally to create clusters, and then sequentially adding fluorescently-labeled reversible terminator nucleotides. After each nucleotide incorporation cycle, a high-resolution camera captures the fluorescent signal from each cluster, identifying the base before the terminator is cleaved to allow the next cycle to begin [105].
For CRISPR validation, the key advantage of NGS is its deep sequencing capability. By sequencing the same genomic locus thousands of times, it can detect and precisely quantify even very rare editing events, with a sensitivity that can reach below 1% allele frequency [106] [104]. This provides an unparalleled, quantitative view of the entire spectrum of mutations in an edited cell population. Furthermore, NGS is capable of multiplexing, where hundreds of samples are barcoded with unique DNA indices, pooled, and sequenced simultaneously, dramatically increasing throughput and reducing per-sample cost for large-scale experiments [106] [102]. The main trade-offs are shorter read lengths (typically 150-300 bp for short-read platforms) and the requirement for sophisticated bioinformatics pipelines to analyze the massive volume of data generated [105] [102].
Head-to-Head Comparison
Table 1: Comparative Analysis of Sanger Sequencing and NGS for CRISPR Validation
| Feature | Sanger Sequencing | Next-Generation Sequencing (NGS) |
|---|---|---|
| Fundamental Method | Chain termination with ddNTPs and capillary electrophoresis [101] [102] | Massively parallel sequencing (e.g., Sequencing by Synthesis) [105] [104] |
| Typical Read Length | Long (500 - 1000 bp), contiguous [103] [102] | Short (50 - 300 bp), fragmented [105] [102] |
| Throughput | Low to medium; one fragment per reaction [104] | Extremely high; millions to billions of fragments per run [105] [102] |
| Detection Sensitivity | ~15-20% allele frequency [104] | <1% allele frequency [106] [104] |
| Quantitative Capability | Low; poor resolution of complex mixtures | High; precise quantification of all alleles present |
| Multiplexing | Not available | High-level; hundreds of samples can be pooled via barcoding [106] |
| Primary Data Output | Single sequence chromatogram | Millions of short reads requiring alignment [102] |
| Bioinformatics Demand | Low; basic sequence analysis tools [101] | High; requires specialized pipelines for alignment and variant calling [102] |
| Best Application in CRISPR | Validation of edits in clonal, homogeneous cell lines or simple models. | Characterization of complex editing outcomes, low-frequency variant detection, and high-throughput screens. |
Experimental Protocols for CRISPR Validation
Sanger Sequencing Validation Workflow
The following protocol outlines the key steps for validating CRISPR edits using Sanger sequencing.
- Step 1: Post-Editing Sample Preparation. Following the CRISPR-Cas9 transfection or electroporation into the target cells, allow sufficient time for the expression of the edited genotype. Harvest genomic DNA from the edited cell population using a standard extraction method. The quality and purity of the genomic DNA are critical for successful PCR amplification.
- Step 2: PCR Amplification of Target Locus. Design primers that flank the CRISPR target site, generating an amplicon of optimal size for Sanger sequencing (typically 500-800 bp). It is critical to use a high-fidelity DNA polymerase to minimize PCR-introduced errors. The primers should be positioned to provide sufficient sequence context around the cut site for clear analysis.
- Step 3: Purification and Sequencing. Purify the resulting PCR product to remove primers, enzymes, and salts. The purified amplicon is then used as the template for a Sanger sequencing reaction, which utilizes a single primer (typically one of the PCR primers) and the chain-termination chemistry. For confidence, sequencing from both the forward and reverse directions is recommended.
- Step 4: Data Analysis. The sequencing results are visualized as a chromatogram. For a clean, homozygous edit, the chromatogram will show a clear, non-overlapping sequence trace with the mutation evident. For a heterogeneous population, the trace will show overlapping peaks starting at the site of the edit. While this confirms editing, the specific indels cannot be deconvoluted and must be inferred or analyzed by other methods, such as cloning and sequencing individual alleles [100] [107].
NGS Validation Workflow (Amplicon Sequencing)
Amplicon sequencing is a targeted NGS approach ideal for high-throughput, deep validation of CRISPR edits.
- Step 1: Library Preparation. Amplify the target locus from the extracted genomic DNA, similar to the Sanger workflow. However, in this method, the PCR primers are designed with overhang adapter sequences that are compatible with the chosen NGS platform (e.g., Illumina). In a subsequent limited-cycle PCR, unique barcode sequences (indexes) are added to each sample's amplicons. This allows for the pooling (multiplexing) of hundreds of different samples in a single sequencing run [106].
- Step 2: Sequencing. The pooled library is loaded onto an NGS flow cell where clusters are generated and sequenced using a platform-specific chemistry like SBS. The process is automated and generates millions of paired-end reads covering the target site from all pooled samples.
- Step 3: Bioinformatics Analysis. The raw data (short reads) undergoes a multi-step computational pipeline:
- Demultiplexing: Reads are assigned to their original sample based on the unique barcode sequences.
- Quality Control & Trimming: Low-quality bases and adapter sequences are removed.
- Alignment: The processed reads are aligned to a reference genome sequence.
- Variant Calling: Specialized algorithms compare the aligned reads to the reference to identify and quantify insertions, deletions, and substitutions at the target site. The output is a comprehensive list of all detected alleles and their frequencies within the sample [106] [108].
Diagram: NGS Amplicon Sequencing Workflow for CRISPR Validation
Decision Framework and Best Practices
Choosing the Right Validation Method
The choice between Sanger and NGS is not a matter of which is universally superior, but which is optimal for the specific experimental context. The following framework aids in this decision:
- Project Scale and Throughput Needs: For projects involving the validation of a few targets (e.g., 1-20) or a small number of samples, Sanger sequencing is highly cost-effective and operationally simple [104]. However, for large-scale projects—such as screening hundreds of sgRNAs, optimizing multiple editing conditions, or conducting genome-wide screens—the multiplexing capability of NGS makes it vastly more efficient and economical on a per-sample basis [106] [102].
- Required Resolution and Sensitivity: If the experimental goal is simply to confirm the presence of an edit in a clonal, homogeneous cell line, Sanger sequencing is often sufficient. Conversely, if the goal is to fully characterize a heterogeneous cell population, quantify editing efficiency, or detect low-frequency editing events (e.g., in a polyclonal pool or for assessing off-target effects), then the deep sequencing and high sensitivity of NGS are indispensable [106] [104].
- Budget and Bioinformatics Considerations: Sanger sequencing has a low barrier to entry in terms of cost and data analysis, requiring minimal bioinformatics expertise. NGS, while offering a lower cost per base for large projects, requires a significant investment in sequencing reagents and, crucially, access to computational resources and personnel skilled in bioinformatics for data analysis and interpretation [102].
The Evolving Role of Sanger in an NGS World
The relationship between Sanger and NGS is increasingly synergistic rather than competitive. A growing body of evidence from clinical genetics suggests that for high-quality NGS variant calls (defined by high depth of coverage and quality scores), orthogonal Sanger validation may be redundant, with studies showing concordance rates of 99.965% to 100% [109] [108]. This challenges the long-held dogma that Sanger validation is always necessary for NGS-discovered variants.
In the context of CRISPR validation, this means that once an NGS pipeline is thoroughly validated and quality thresholds are established, Sanger's role shifts. It remains invaluable for spot-checking key results, troubleshooting failed NGS runs, or validating specific edits in clonal lines isolated from a larger, NGS-characterized pool. Its simplicity and speed make it perfect for these targeted confirmations, even as NGS handles the heavy lifting of primary discovery and quantification.
Essential Reagents and Tools
Table 2: The Scientist's Toolkit for CRISPR Genotyping Validation
| Reagent / Solution | Function in Validation Workflow |
|---|---|
| High-Fidelity DNA Polymerase | Accurate amplification of the target locus from genomic DNA, minimizing PCR errors that could be mistaken for real edits [100]. |
| NGS Library Preparation Kit | Commercial kits provide the enzymes, buffers, and adapters needed to convert PCR amplicons into a sequencing-ready library [106]. |
| DNA Barcodes/Indexes | Short, unique DNA sequences added to each sample's amplicons during PCR, enabling multiplexing of hundreds of samples in one NGS run [106]. |
| Sanger Sequencing Primers | Oligonucleotides designed to bind adjacent to the target site to initiate the sequencing reaction. Specificity is critical [100] [108]. |
| Bioinformatics Software | Tools for NGS data analysis, including aligners (e.g., BWA, NovoAlign) and variant callers (e.g., GATK), are essential for interpreting results [109] [108]. |
| Cloning Vector Systems | Used in conjunction with Sanger sequencing to isolate and sequence individual alleles from a mixed population, overcoming its deconvolution limitation [100]. |
Diagram: Method Selection Decision Tree
The accurate genotyping of CRISPR-Cas9 editing outcomes is a non-negotiable pillar of rigorous research. As this guide outlines, both Sanger sequencing and Next-Generation Sequencing offer powerful, complementary pathways to this end. Sanger remains the unrivaled method for fast, cost-effective validation of simple edits, while NGS provides a deep, quantitative view of editing complexity at scale. The optimal validation strategy is not a binary choice but a deliberate one, informed by experimental goals, project scale, and required resolution. By applying the principles and protocols detailed herein, researchers can ensure the accuracy and reliability of their CRISPR gene editing research, forming a solid foundation for scientific discovery and therapeutic development.
Gene editing has become a cornerstone of modern molecular biology, enabling precise modifications to an organism's DNA to investigate gene function, develop therapeutic interventions, and create genetically modified organisms for agricultural applications [110]. The field has evolved significantly from early homologous recombination techniques to programmable nucleases that can target specific genomic sequences with remarkable accuracy. Three major technologies have dominated this landscape: Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and the more recent Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) system [110] [111]. Understanding the comparative advantages and limitations of these platforms is essential for researchers selecting the appropriate tool for their specific applications. This technical guide provides a comprehensive comparison of these three gene editing technologies, focusing on their precision, cost, and ease of use within the context of basic principles of CRISPR gene editing research.
Zinc Finger Nucleases (ZFNs)
ZFNs represent one of the earliest programmable gene editing technologies. These engineered proteins function by combining a zinc finger DNA-binding domain with the FokI nuclease domain [110] [111]. Each zinc finger domain recognizes a specific DNA triplet, and multiple domains must be assembled to target a unique sequence [110]. Once the zinc finger array binds to the target DNA, the FokI nuclease domain induces a double-strand break (DSB) [111]. The cellular repair mechanisms, primarily non-homologous end joining (NHEJ) or homology-directed repair (HDR), are then activated to introduce genetic modifications at the break site [111].
Transcription Activator-Like Effector Nucleases (TALENs)
TALENs operate on a similar principle to ZFNs but utilize transcription activator-like effector (TALE) proteins for DNA recognition [110] [111]. Each TALE repeat recognizes a single nucleotide, offering greater flexibility and precision than ZFNs [110] [111]. Like ZFNs, TALENs are fused to the FokI nuclease domain to create targeted double-strand breaks after DNA binding [111]. The one-to-one correspondence between TALE repeats and nucleotides simplifies design compared to ZFNs, though the assembly process remains technically challenging [111].
CRISPR-Cas Systems
The CRISPR-Cas system, derived from a bacterial adaptive immune system, represents a fundamentally different approach to gene editing [110]. The most commonly used CRISPR system employs a guide RNA (gRNA) to direct the Cas9 nuclease to complementary DNA sequences [110] [111]. The gRNA can be easily programmed to target different genomic loci without requiring protein engineering [110]. For successful target recognition and cleavage, the CRISPR-Cas9 system requires a protospacer adjacent motif (PAM) sequence adjacent to the target site [111]. Once bound to the target DNA, Cas9 creates a double-strand break, triggering the cell's natural DNA repair mechanisms [110].
Figure 1: Molecular Mechanisms of Major Gene Editing Technologies. Each system uses different targeting approaches but converges on creating double-strand breaks (DSBs) that trigger cellular DNA repair mechanisms.
Comparative Performance Analysis
Quantitative Comparison of Key Parameters
The following table summarizes the direct comparison between CRISPR, ZFNs, and TALENs across critical performance metrics based on current research and implementation data:
Table 1: Comprehensive Feature Comparison of Gene Editing Platforms
| Feature | CRISPR | ZFN | TALEN |
|---|---|---|---|
| Targeting Mechanism | RNA-guided (gRNA) | Protein-based (Zinc fingers) | Protein-based (TALE repeats) |
| Target Recognition | 17-24 nt guide sequence | DNA triplet per zinc finger | Single nucleotide per TALE repeat |
| Nuclease Component | Cas9 | FokI dimer | FokI dimer |
| Precision | Moderate to high [110] | High [110] | High [110] |
| Off-Target Effects | Higher potential [110] | Lower potential [111] | Lowest potential [111] |
| Ease of Design | Simple (days) [110] | Complex (months) [110] [111] | Moderate (weeks) [111] |
| Cost | Low [110] | High [110] | High [110] |
| Scalability | High (ideal for high-throughput) [110] | Limited [110] | Limited [110] |
| Multiplexing Capacity | High (multiple gRNAs) [110] | Low | Low |
| Time Required for Workflow | 3 months (knockouts) [112] | Several months [111] | Several weeks [111] |
| Delivery Methods | Viral vectors, nanoparticles [110] | Primarily plasmid vectors [110] | Plasmid vectors |
Experimental Workflow and Protocol Considerations
The practical implementation of gene editing technologies follows a structured workflow from design to validation. CRISPR experiments typically require 3 months for knockout generation and 6 months for knock-ins, with researchers reporting repeating the entire workflow approximately 3 times before success [112]. The difficulty of editing varies significantly by cell type, with primary cells (like T-cells) proving more challenging than immortalized cell lines [112].
Table 2: Experimental Workflow Comparison
| Workflow Stage | CRISPR | ZFN | TALEN |
|---|---|---|---|
| Target Design | gRNA selection based on PAM site [111] | Zinc finger assembly for triplets [111] | TALE repeat assembly for nucleotides [111] |
| Component Engineering | gRNA synthesis (days) [110] | Protein engineering (months) [110] [111] | Protein engineering (weeks) [111] |
| Delivery | Viral vectors, LNPs, electroporation [110] | Plasmid vectors, mRNA [110] | Plasmid vectors, mRNA |
| Validation Timeline | 2-4 weeks | 4-8 weeks | 4-8 weeks |
| Difficulty in Primary Cells | High (50% find difficult) [112] | High | High |
Figure 2: Comparative Experimental Workflow for Gene Editing Technologies. While all platforms share common stages, the engineering phase differs significantly in complexity and duration.
Research Reagent Solutions and Essential Materials
Successful implementation of gene editing technologies requires specific reagent systems and components tailored to each platform:
Table 3: Essential Research Reagents for Gene Editing Platforms
| Reagent Category | CRISPR-Specific | ZFN-Specific | TALEN-Specific |
|---|---|---|---|
| Nuclease Components | Cas9, Cas12, Cas13 proteins [113] | Zinc finger-FokI fusion proteins | TALE repeat-FokI fusion proteins |
| Targeting Molecules | Guide RNA (gRNA) libraries [114] | Zinc finger arrays | TALE repeat arrays |
| Delivery Systems | Lipid nanoparticles (LNPs) [26], Viral vectors | Plasmid vectors, mRNA [110] | Plasmid vectors |
| Validation Tools | NGS-based off-target assays, ICE analysis [112] | Specificity validation assays | Specificity validation assays |
| Commercial Kits | CRISPR kits & reagents [114] [115] | ZFN engineering kits | TALEN assembly kits |
Current Applications and Clinical Translation
Therapeutic Applications and Clinical Trials
The gene editing landscape has witnessed remarkable clinical advancements, particularly for CRISPR-based therapies. As of February 2025, approximately 250 clinical trials involve gene-editing therapeutic candidates, with over 150 trials currently active [116]. The first CRISPR-based medicine, CASGEVY (exagamglogene autotemcel), received regulatory approval for sickle cell disease (SCD) and transfusion-dependent beta thalassemia (TBT) [113] [26]. This milestone demonstrated CRISPR's therapeutic potential and paved the way for numerous other clinical applications.
Current clinical trials span multiple therapeutic areas, with blood disorders continuing to lead the field [116]. Phase 3 trials are also underway in hereditary amyloidosis and immunodeficiencies [116]. Notable advancements include Intellia Therapeutics' phase I trial for hereditary transthyretin amyloidosis (hATTR), the first clinical trial for a CRISPR-Cas9 therapy delivered by lipid nanoparticle (LNP) [26]. This trial demonstrated rapid, deep, and long-lasting reductions in disease-related protein levels, with sustained response through two years of follow-up [26].
Agricultural and Industrial Applications
Beyond therapeutic applications, gene editing technologies have shown significant promise in agricultural and industrial sectors. CRISPR in particular has enabled precise modifications to enhance crop yield, nutritional quality, and stress resistance [111] [117]. The technology allows development of crops with improved characteristics without introducing foreign DNA, potentially streamlining regulatory processes [117]. Agricultural applications include creating disease-resistant crops, enhancing photosynthetic efficiency, and improving shelf life, taste, and texture [113] [111].
Technical Challenges and Limitations
Platform-Specific Technical Hurdles
Each gene editing platform faces distinct technical challenges that influence their suitability for different applications:
CRISPR-Specific Challenges: Off-target effects remain a primary concern, though improved Cas enzymes like high-fidelity Cas9 are addressing this limitation [110]. Immune responses against bacterial-derived Cas proteins in human patients present another challenge for therapeutic applications [110]. Delivery efficiency to target tissues and cells also varies significantly, with lipid nanoparticles showing particular promise for liver-targeted therapies [26].
ZFN Limitations: The complexity of design and requirement for extensive protein engineering restrict ZFNs' accessibility and scalability [110] [111]. The limited target range (approximately 18 base pairs) and potential cytotoxicity in some applications further constrain their utility [111]. The high development cost and technical expertise required make ZFNs less practical for widespread adoption [110].
TALEN Constraints: While offering high specificity, TALENs present delivery challenges due to their large size (typically 2 kb larger than ZFNs) [111]. Longer TALEN constructs show decreased accuracy, creating design limitations [111]. The repetitive nature of TALEN sequences can also complicate cloning and viral packaging [111].
Shared Challenges Across Platforms
All gene editing technologies face common obstacles including efficient delivery to target cells, managing potential off-target effects, and ensuring comprehensive safety profiles [110]. The high cost of therapy development and manufacturing presents another significant barrier, with estimates exceeding $1 billion to bring a single gene therapy to market [117]. Ethical considerations and evolving regulatory frameworks also impact all gene editing platforms, particularly regarding germline modifications and appropriate therapeutic applications [110] [117].
Future Directions and Emerging Innovations
Next-Generation CRISPR Technologies
The CRISPR landscape continues to evolve with several advanced systems addressing current limitations:
Base Editing: This technology enables direct, precise conversion of one DNA base to another without creating double-strand breaks, significantly reducing off-target risks [110]. Base editors combine a catalytically impaired Cas protein with a deaminase enzyme, offering higher precision for therapeutic applications requiring single-nucleotide changes.
Prime Editing: This more recent innovation allows for targeted insertions, deletions, and all possible base-to-base conversions without double-strand breaks [110]. Prime editing uses a catalytically impaired Cas9 fused to a reverse transcriptase and a prime editing guide RNA (pegRNA), dramatically expanding the scope of possible genetic modifications.
Novel Cas Variants: The discovery and engineering of additional Cas proteins (Cas12, Cas13, and others) continue to broaden CRISPR applications beyond DNA editing to include RNA targeting [110]. Ultracompact Cas variants developed by companies like Mammoth Biosciences enable more efficient delivery using smaller viral vectors [113].
Market Trends and Adoption Trajectory
The CRISPR-based gene editing market is projected to grow from USD 7.06 billion in 2025 to approximately USD 24.37 billion by 2034, representing a compound annual growth rate (CAGR) of 14.76% [113]. This growth significantly outpaces the broader gene editing market, expected to grow at a CAGR of 10.2% from 2025 to 2032 [114]. North America currently dominates the market with a 41.88% revenue share in 2024, though the Asia-Pacific region is anticipated to witness the fastest growth [113].
The pharmaceutical and biotechnology sectors represent the largest end-users of CRISPR technology [115], with agricultural applications showing the most rapid growth potential [117]. The services segment is also expanding rapidly as researchers increasingly outsource gene editing to specialized providers [113]. These trends indicate continued diversification and maturation of CRISPR applications across multiple sectors.
The comparative analysis of CRISPR, ZFNs, and TALENs reveals a complex landscape where each technology offers distinct advantages for specific applications. CRISPR excels in ease of use, cost-effectiveness, and scalability, making it ideal for high-throughput experiments and applications requiring multiplexed editing. ZFNs and TALENs maintain relevance for projects demanding exceptionally high precision and validated specificity, particularly in therapeutic contexts where their longer development history provides regulatory advantages.
The selection of an appropriate gene editing platform ultimately depends on research goals, required precision, available resources, and specific application requirements. While CRISPR currently dominates the field in terms of adoption rate and commercial growth, all three technologies continue to evolve with improvements in specificity, delivery, and applications. As innovation progresses, researchers can anticipate even more refined tools that will further expand the possibilities for precise genetic engineering across basic research, therapeutic development, and agricultural applications.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas system has revolutionized biological science, providing an unprecedented ability to modify genes for research, therapeutic, and agricultural applications [110]. However, the therapeutic promise of CRISPR gene editing can only be realized if its molecular components—the Cas nuclease and guide RNA (gRNA)—can be efficiently, safely, and precisely delivered to target cells [45] [118]. Delivery remains a fundamental challenge in CRISPR-based applications, as these macromolecular complexes cannot passively cross cell membranes and require specialized vehicles for cellular entry [119] [120]. The ideal delivery system must protect CRISPR cargo from degradation, facilitate cellular uptake, enable endosomal escape, minimize off-target effects, and avoid immune activation [45] [121]. This technical evaluation examines the efficiency and toxicity profiles of current CRISPR delivery platforms, providing researchers with a comparative framework for method selection within the broader context of basic CRISPR gene editing principles.
Different delivery approaches present distinct trade-offs between editing efficiency, specificity, cargo capacity, and safety [120]. Viral vectors leverage natural infection mechanisms but can provoke immune responses, while non-viral methods like lipid nanoparticles offer favorable safety profiles but variable efficiency [45] [118]. The choice of CRISPR cargo format—DNA, mRNA, or ribonucleoprotein (RNP)—further influences these dynamics, affecting editing kinetics, duration of Cas9 activity, and potential for off-target effects [120]. This guide systematically evaluates these dimensions to inform selection of appropriate delivery strategies for specific research or therapeutic objectives.
CRISPR Cargo Formats
The molecular format of CRISPR components significantly influences editing precision, kinetics, and delivery requirements. The three primary cargo formats each present distinct advantages and limitations for genome editing applications [120].
DNA Plasmid Delivery
Plasmid-based delivery involves introducing DNA sequences encoding both the Cas9 nuclease and gRNA into target cells. This approach represents the simplest and most cost-effective method, with a single plasmid potentially containing all necessary components for gene editing [120]. However, plasmid delivery requires both transcription and translation before active editing can occur, resulting in delayed onset of nuclease activity. More critically, prolonged persistence and expression of CRISPR components in cells can lead to increased off-target effects due to sustained Cas9 activity [120]. There is also a risk of insertional mutagenesis if plasmid DNA integrates randomly into the host genome. While inducible systems such as tetracycline-responsive promoters can help control the timing of Cas9 expression, these concerns have limited the clinical translation of plasmid-based approaches [120].
mRNA Delivery
Delivery of in vitro transcribed mRNA encoding the Cas9 nuclease, combined with a separate gRNA, bypasses the transcription step required for DNA-based approaches [120]. This enables more rapid translation of Cas9 protein and consequently faster onset of editing activity. The inherent instability of mRNA leads to transient expression of CRISPR components, reducing the duration of Cas9 exposure and potentially minimizing off-target effects. mRNA delivery also eliminates the risk of genomic integration associated with DNA-based approaches. However, mRNA-based delivery presents manufacturing challenges, as production is more expensive and technically complex compared to plasmid DNA [120]. The inherent instability of mRNA also requires careful handling and optimized formulation to maintain integrity during delivery.
Ribonucleoprotein (RNP) Delivery
RNP delivery involves direct introduction of preassembled complexes of Cas9 protein and gRNA into target cells [120]. This approach provides the most rapid editing activity, as the functional complex can immediately localize to the nucleus and begin genome editing without any intermediate steps. The transient nature of RNP activity—limited by protein turnover—minimizes off-target effects and eliminates risks of genomic integration. RNP delivery has demonstrated notable clinical success, with the first FDA-approved CRISPR therapy, Casgevy for sickle cell anemia, utilizing this approach [120]. However, RNPs present handling challenges due to their susceptibility to protease degradation and require more labor-intensive, expensive production processes compared to nucleic acid-based formats [120].
Table 1: Comparison of CRISPR Cargo Formats
| Cargo Format | Editing Kinetics | Specificity | Safety Considerations | Manufacturing Complexity |
|---|---|---|---|---|
| DNA Plasmid | Slow (requires transcription and translation) | Lower (prolonged Cas9 expression increases off-target risk) | Risk of insertional mutagenesis; bacterial sequence-related immunogenicity | Low; cost-effective and amenable to scaling |
| mRNA | Moderate (requires translation only) | Moderate (transient expression limits exposure) | No genomic integration risk; mRNA immunogenicity possible | Moderate; cell-free production but technical challenges in large-scale manufacturing |
| RNP | Fast (immediately active) | High (transient activity minimizes off-target effects) | No genomic integration risk; reduced immunogenicity compared to viral delivery | High; labor-intensive production with risk of toxic contaminants |
Delivery Platforms: Mechanisms and Characteristics
Viral Vector Systems
Viral vectors exploit the natural efficiency of viral infection mechanisms to deliver CRISPR components to target cells. These systems are particularly valuable for hard-to-transfect cells and in vivo applications [120].
Adeno-Associated Viral Vectors (AAVs) are small, non-pathogenic viruses that have become one of the most popular delivery methods for CRISPR applications, particularly in clinical settings [45]. AAVs offer several advantages, including mild immune responses, non-integration into the host genome (reducing mutagenesis risk), and FDA approval for certain gene therapies. However, AAVs are severely limited by their small cargo capacity (~4.7 kb), which is insufficient for standard SpCas9 (4.2 kb) plus gRNAs and repair templates [45] [120]. Strategies to overcome this limitation include using smaller Cas9 orthologs (e.g., SaCas9), splitting components across multiple vectors, or employing dual AAV systems [45] [120]. AAVs also offer tissue targeting capabilities through different serotypes and engineered capsids [120].
Lentiviral Vectors (LVs) are retroviral vectors derived from HIV that efficiently integrate into the host genome, enabling long-term expression of CRISPR components [45] [120]. This characteristic makes LVs particularly valuable for applications requiring persistent Cas9 expression, such as genetic screens using CRISPR libraries. LVs can deliver large cargo sequences and infect both dividing and non-dividing cells. However, the integrating nature of LVs raises significant safety concerns regarding insertional mutagenesis, potentially disrupting tumor suppressor genes or activating oncogenes [45] [120]. While integrase-deficient lentiviral vectors (IDLVs) reduce integration rates, they still present some risk and generally exhibit lower transduction efficiency [120].
Adenoviral Vectors (AdVs) are non-integrating viral vectors with a large cargo capacity (up to 36 kb), enabling delivery of full CRISPR systems with repair templates [45]. AdVs can infect diverse cell types and be grown to high titers. However, their common occurrence in human populations means many individuals have pre-existing immunity, potentially leading to strong immune responses and rapid vector clearance [45]. This immunogenicity has limited their clinical application compared to AAVs.
Non-Viral Delivery Systems
Non-viral delivery methods typically use synthetic or biological materials to package and deliver CRISPR components, offering favorable safety profiles but variable efficiency [45] [118].
Lipid Nanoparticles (LNPs) are synthetic nanoparticles composed of ionizable lipids that encapsulate and protect CRISPR cargo [45]. LNPs gained prominence through their use in COVID-19 mRNA vaccines and have since been adapted for CRISPR delivery. They offer minimal safety concerns due to lack of viral components and can deliver all three cargo formats (DNA, mRNA, RNP) [45] [120]. A significant advancement reported in 2025 involves lipid nanoparticle spherical nucleic acids (LNP-SNAs), which wrap CRISPR tools in a protective DNA shell [119]. This architecture enhances cellular uptake and improves endosomal escape, addressing a key limitation of traditional LNPs. In testing, LNP-SNAs demonstrated three times greater cell entry, tripled gene-editing efficiency, and reduced toxicity compared to standard delivery systems [119]. However, LNP efficiency varies considerably across cell types, and they generally show lower transfection efficiency compared to viral methods or electroporation [120].
Extracellular Vesicles (EVs) are natural, cell-derived lipid envelopes that transport bioactive molecules between cells [45] [118]. As delivery vehicles, EVs offer inherent biocompatibility and potential for tissue-specific homing. Their natural composition reduces immunogenicity concerns compared to synthetic systems. However, clinical translation of EV-based delivery has been hindered by manufacturing challenges related to their heterogeneity and complexity [45]. Standardizing production and loading methods for EVs remains an active area of research.
Electroporation uses electrical pulses to create temporary pores in cell membranes, allowing direct passage of CRISPR components into the cytoplasm [120]. This physical method achieves high efficiency, particularly for RNP delivery, and works across diverse cell types. However, electroporation can cause significant cell damage and death, requiring careful optimization of electrical parameters [120]. It is primarily suitable for ex vivo applications, such as the editing of hematopoietic stem cells for Casgevy therapy [120].
Table 2: Platform Efficiency and Toxicity Profiles
| Delivery Method | Theoretical Efficiency | Practical Efficiency | Toxicity Concerns | Cargo Capacity | Best Applications |
|---|---|---|---|---|---|
| AAV | High (natural tropism) | Moderate (limited by cargo size) | Low immunogenicity, mild immune responses | Low (~4.7 kb) | In vivo delivery, clinical therapy |
| Lentivirus | High (integrating) | High | Insertional mutagenesis, moderate immunogenicity | High (~8 kb) | In vitro and ex vivo editing, CRISPR screens |
| Adenovirus | Moderate | Moderate | High immunogenicity, inflammatory responses | Very high (~36 kb) | In vivo delivery requiring large cargo |
| LNP | Variable | Low to moderate (cell-type dependent) | Low toxicity, minimal immunogenicity | Moderate | In vivo mRNA/RNP delivery, clinical applications |
| LNP-SNA | High (enhanced uptake) | High (3× standard LNP) | Reduced toxicity reported | Moderate | Potential therapeutic applications [119] |
| Electroporation | High | High (especially for RNP) | High (cell damage and death) | No practical limit | Ex vivo editing (e.g., blood cells, stem cells) |
Quantitative Comparison of Delivery Performance
Direct comparison of delivery platforms reveals significant differences in performance metrics. Recent studies have quantified these variations to provide guidance for method selection.
Table 3: Quantitative Performance Metrics Across Delivery Systems
| Delivery Platform | Editing Efficiency Range | Cell Viability | Off-Target Rate (Relative) | Duration of Expression |
|---|---|---|---|---|
| AAV | 40-70% (varies by serotype and tissue) | High (>85%) | Low to moderate | Long-term (months to years) |
| Lentivirus | 60-90% in permissive cells | Moderate (70-85%) | High (due to persistent expression) | Long-term (stable integration) |
| Adenovirus | 30-60% | Moderate (60-80%) | Moderate | Medium-term (weeks) |
| Standard LNP | 20-50% (highly variable) | High (>80%) | Low to moderate | Transient (days) |
| LNP-SNA | 60-90% (3× improvement reported) | High (>90%, reduced toxicity) | Lower precision (60% improvement in precise repair) | Transient (days) [119] |
| Electroporation (RNP) | 70-95% | Low to moderate (40-70%) | Lowest | Shortest (hours to days) |
The LNP-SNA platform represents a particularly significant advancement in non-viral delivery. In addition to tripling gene-editing efficiency compared to standard lipid nanoparticles, this system improved the success rate of precise DNA repairs by more than 60% while demonstrating reduced cellular toxicity [119]. The spherical nucleic acid architecture facilitates enhanced cellular uptake through interactions with cell surface receptors, enabling more efficient delivery to the nucleus where editing occurs [119].
Experimental Protocols for Delivery Evaluation
Protocol: Evaluating LNP-SNA Delivery Efficiency
This protocol adapts methodology from recent studies demonstrating enhanced CRISPR delivery using spherical nucleic acid structures [119].
Materials:
- CRISPR-Cas9 components (Cas9 mRNA, sgRNA, DNA repair template if using HDR)
- Lipid nanoparticles (LNPs) and functionalization reagents for SNA construction
- Target cells (e.g., human bone marrow stem cells, keratinocytes, or other relevant cell types)
- Transfection efficiency controls (e.g., fluorescent reporter plasmids)
- Cell culture media and supplements
- Genomic DNA extraction kit
- PCR reagents and sequencing primers
- T7E1 assay or TIDE analysis reagents for editing efficiency quantification
Procedure:
- Synthesize LNP-SNAs by formulating LNPs with CRISPR cargo (mRNA or RNP format) then applying a dense shell of oligonucleotides to create the spherical nucleic acid architecture [119].
- Culture target cells in appropriate medium until 60-70% confluent.
- Treat cells with LNP-SNAs containing CRISPR components; include controls:
- Standard LNP with same CRISPR cargo
- Untreated cells
- Fluorescent transfection control (e.g., GFP mRNA)
- Incubate for 48-72 hours to allow editing to occur.
- Harvest cells and extract genomic DNA using commercial kits.
- Amplify target region by PCR using flanking primers.
- Quantify editing efficiency using next-generation sequencing, T7E1 assay, or TIDE analysis.
- Assess cell viability using MTT or CellTiter-Glo assays alongside editing assessment.
- Evaluate off-target effects by examining predicted off-target sites using targeted sequencing.
Expected Outcomes: LNP-SNAs should demonstrate significantly higher editing efficiency (typically 2-3× improvement) and reduced cytotoxicity compared to standard LNPs [119].
Protocol: Electroporation of CRISPR RNPs in Primary Cells
This protocol outlines optimal conditions for delivering CRISPR components as ribonucleoproteins via electroporation, based on methods used for therapeutic applications like Casgevy [120].
Materials:
- Purified Cas9 protein
- Synthetic sgRNA
- Electroporation system (e.g., Neon or Amaxa systems)
- Electroporation buffers
- Primary cells (e.g., T cells, hematopoietic stem cells)
- Cell culture media with appropriate cytokines and supplements
- Genomic DNA extraction kit
- Editing efficiency analysis reagents
Procedure:
- Precomplex RNPs by incubating Cas9 protein with sgRNA at molar ratio of 1:1.2 for 10-20 minutes at room temperature.
- Harvest and wash cells to remove serum and ions that may interfere with electroporation.
- Resuspend cells in appropriate electroporation buffer at high density (e.g., 1-10 × 10⁶ cells/mL).
- Mix cells with RNP complexes and transfer to electroporation cuvette.
- Electroporate using device-specific parameters optimized for cell type:
- For T cells: 1600V, 10ms, 3 pulses (Neon system)
- For HSCs: 1400V, 20ms, 1 pulse (Neon system)
- Immediately transfer cells to pre-warmed culture medium.
- Assess viability 24 hours post-electroporation using trypan blue exclusion or flow cytometry with viability dyes.
- Culture cells for 3-7 days to allow expression of editing outcomes.
- Harvest cells and analyze editing efficiency as described in Protocol 5.1.
Troubleshooting: Low viability may require optimization of pulse parameters or cell density. Low editing efficiency may indicate RNP degradation or suboptimal sgRNA design.
Emerging Technologies and Future Directions
AI-Designed CRISPR Systems
Recent advances in artificial intelligence have enabled the design of novel CRISPR systems with enhanced properties. In 2025, researchers used large language models trained on 1 million CRISPR operons to generate OpenCRISPR-1, an AI-designed editor that shows comparable or improved activity and specificity relative to SpCas9 despite being 400 mutations away in sequence [44]. This approach represents a promising alternative to natural Cas9 orthologs, potentially overcoming functional tradeoffs observed when microbial systems are ported into human cells.
Advanced Nanoparticle Systems
The development of LNP-SNAs demonstrates how structural innovations in nanomedicine can enhance delivery efficiency without changing core components [119]. These DNA-coated nanostructures supercharge delivery by facilitating receptor-mediated cellular uptake and improving endosomal escape. The modular nature of this platform allows adaptation for various therapeutic applications, with ongoing research focused on tissue-specific targeting [119].
Virus-Like Particles (VLPs)
VLPs represent a hybrid approach that combines advantages of viral and non-viral delivery. These engineered particles consist of empty viral capsids lacking viral genetic material, making them non-replicative and non-integrating [45]. VLPs can be designed for cell-specific delivery and demonstrate improved endosomal escape compared to synthetic nanoparticles. While manufacturing challenges and stability issues have hindered clinical translation, VLPs offer a promising direction for achieving efficient delivery with enhanced safety profiles [45].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Delivery Evaluation
| Reagent/Category | Function | Examples/Sources |
|---|---|---|
| Transfection Controls | Validate delivery efficiency independent of editing | GFP mRNA/reporter plasmids [122] |
| Positive Editing Controls | Verify system functionality with validated gRNAs | gRNAs targeting TRAC, RELA, CDC42BPB (human); ROSA26 (mouse) [122] |
| Negative Editing Controls | Establish baseline for non-specific effects | Scramble gRNAs, guide-only, Cas9-only [122] |
| Viability Assays | Quantify delivery-associated toxicity | MTT, CellTiter-Glo, Annexin V staining |
| Editing Validation | Assess on-target efficiency | T7E1 assay, TIDE analysis, next-generation sequencing [122] |
| Off-Target Assessment | Evaluate specificity | GUIDE-seq, CIRCLE-seq, targeted sequencing [121] |
The selection of appropriate delivery methods represents a critical decision point in CRISPR experimental design, with significant implications for editing efficiency, specificity, and practical feasibility. Viral vectors, particularly AAVs and lentiviruses, offer high efficiency but present safety concerns including immunogenicity and insertional mutagenesis [45] [120]. Non-viral methods like LNPs and electroporation provide favorable safety profiles but variable efficiency, though recent advancements in LNP-SNAs demonstrate substantial improvements in delivery performance [119]. The choice of cargo format further influences experimental outcomes, with RNP delivery offering superior specificity through transient activity while plasmid DNA provides cost-effective but less precise alternatives [120].
As CRISPR technology continues evolving, emerging approaches including AI-designed editors [44] and advanced nanoparticle systems [119] promise to overcome current limitations in delivery efficiency and specificity. By carefully matching delivery strategies to experimental requirements and rigorously validating outcomes with appropriate controls [122], researchers can maximize the potential of CRISPR technologies across diverse applications from basic research to therapeutic development.
Regulatory and Safety Landscape for Clinical CRISPR Applications
The transition of CRISPR-based genome editing from a powerful research tool to a clinical reality represents a paradigm shift in therapeutic development. Framed within the broader thesis of CRISPR's basic principles—which encompass programmable RNA-guided nucleases, DNA repair mechanisms, and delivery systems—this guide examines the evolving regulatory and safety considerations for clinical applications. Since the initial discovery of CRISPR-Cas systems as adaptive immune mechanisms in bacteria, the technology has undergone rapid refinement, yielding increasingly precise editors like base and prime editors that expand the therapeutic scope beyond initial Cas9 systems [123] [52]. The first regulatory approvals of CRISPR-based medicines, such as Casgevy for sickle cell disease and transfusion-dependent beta thalassemia, have established a foundational precedent, prompting the development of tailored regulatory pathways for these innovative products [26] [124].
This technical guide synthesizes the current regulatory requirements and safety assessment methodologies for CRISPR clinical applications, providing researchers and drug development professionals with actionable frameworks for navigating this complex landscape. As the field progresses toward more sophisticated in vivo applications and personalized approaches, understanding these parameters becomes critical for the successful translation of CRISPR technologies into safe and effective human therapies.
Current Regulatory Frameworks and Adaptive Pathways
Evolution from Product-Specific to Platform-Based Regulation
Traditional regulatory pathways for genetic medicines were designed for products with fixed manufacturing specifications and consistent final product composition. However, the emerging paradigm of "CRISPR platformization" represents a significant shift toward regulatory acceptance of platform-based approaches where the core editing system remains constant, with only minimal modifications (such as guide RNA sequences) to target different mutations [125]. This framework enables the development of "umbrella trials" that enroll patients with the same clinical syndrome caused by different genetic mutations, all under a single master protocol [125].
The U.S. Food and Drug Administration (FDA) has recently proposed a "plausible mechanism" pathway for approving on-demand gene editing medicines. This approach considers granting approval when two key conditions are met: (1) the medicine treats patients with the same clinical syndrome regardless of the specific underlying mutation, and (2) it demonstrates consistent patient-to-patient efficacy of a scale that cannot be expected under standard care [125]. This regulatory innovation is particularly crucial for rare genetic diseases, where the commercial viability of developing individual therapies for each unique mutation is limited.
Regulatory Pathways for Different CRISPR Modalities
The regulatory considerations vary significantly based on the editing approach and delivery method. The following table summarizes the key regulatory and safety features of major CRISPR therapeutic platforms:
Table: Regulatory and Safety Profiles of Major CRISPR Therapeutic Platforms
| Platform | Editing Mechanism | Primary Safety Considerations | Regulatory Stage | Example Therapeutics |
|---|---|---|---|---|
| CRISPR-Cas9 | Double-strand break followed by NHEJ or HDR | Off-target effects, chromosomal rearrangements, immunogenicity | Approved therapies; multiple Phase III trials | Casgevy (exa-cel), NTLA-2001 (nex-z) [26] [3] |
| Base Editors | Chemical conversion of single DNA bases without double-strand breaks | Off-target editing, bystander edits, partial efficiency | Phase I/II trials, first clinical data emerging | VERVE-101, VERVE-102 for cardiovascular disease [3] [126] |
| Prime Editors | Reverse transcription of desired edit from pegRNA template | Off-target integration, efficiency challenges | First ex vivo clinical trials initiated | PM359 for chronic granulomatous disease [127] [126] |
| Epigenetic Editors | Modulation of gene expression without altering DNA sequence | Durability of effect, off-target chromatin modifications | Early-phase trials in specific indications | CRISPR-based therapy for facioscapulohumeral muscular dystrophy [126] |
Regulatory Pathway Visualization
The following diagram illustrates the key decision points in the current regulatory pathway for CRISPR-based therapeutics:
Safety Assessment Methodologies and Risk Mitigation
Comprehensive Off-Target Analysis
Off-target editing remains a primary safety concern for CRISPR therapeutics, as unintended modifications in genomic regions with sequence similarity to the target site could potentially disrupt normal gene function or regulation. The field has developed increasingly sophisticated methodologies to identify and quantify these events:
Computational Prediction and In Silico Tools: Initial off-target assessment typically begins with bioinformatic prediction using tools that identify genomic sites with high sequence similarity to the guide RNA target sequence, including mismatches and bulges. Recent advances incorporate machine learning algorithms, with studies showing that RNN-GRU, 5-layer feedforward neural networks, and MLP variants provide optimal prediction results [75]. A similarity-based pre-evaluation methodology using cosine, Euclidean, and Manhattan distance metrics has been developed to identify optimal source datasets for transfer learning in CRISPR-Cas9 off-target prediction [75].
Experimental Validation Methods: Several robust experimental techniques have been developed for empirical off-target detection:
- DISCOVER-Seq and AutoDISCO: These methods leverage the cellular DNA repair machinery to identify off-target sites. AutoDISCO represents an advanced adaptation that enables clinically feasible off-target detection using minimal patient tissue by refining DISCOVER-Seq with cancer research reagents, creating a faster, scalable method that meets regulatory demands [75].
- GUIDE-Seq: This approach uses double-stranded oligodeoxynucleotides to tag double-strand break sites for subsequent sequencing and identification.
- CIRCLE-Seq: An in vitro method that uses purified genomic DNA and CRISPR ribonucleoproteins to identify potential off-target sites in a controlled environment.
The following table outlines essential reagents and their functions in safety assessment protocols:
Table: Essential Research Reagents for CRISPR Safety Assessment
| Reagent/Category | Function in Safety Assessment | Specific Examples/Applications |
|---|---|---|
| CRISPR-Cas Ribonucleoproteins | In vitro cleavage assays to predict specificity | SpCas9, SaCas9, high-fidelity variants (eSpCas9, SpCas9-HF1) [123] |
| gRNA Libraries | Genome-wide off-target screening | Focused libraries for predicted off-target sites; genome-wide libraries for unbiased discovery |
| DNA Repair Biomarkers | Detection of double-strand breaks | Antibodies against γH2AX, 53BP1; used in immunofluorescence assays |
| Cell Line Panels | Assessment in diverse genetic contexts | iPSCs, primary cells, cell lines with varying genetic backgrounds |
| In Vivo Model Systems | Evaluation of off-target effects in physiological contexts | Mouse models, organoids, xenograft systems |
| Sequencing Reagents | Comprehensive genomic analysis | Whole-genome sequencing, targeted amplicon sequencing |
Delivery System Safety Profiles
The choice of delivery system significantly influences the safety profile of CRISPR therapeutics, with distinct considerations for viral versus non-viral approaches:
Viral Vector Systems:
- Adeno-Associated Viruses (AAVs): Widely used for in vivo delivery due to high transduction efficiency, but limited by cargo size constraints (~4.7 kb) and potential immunogenicity. Safety concerns include possible integration of vector genomes into Cas9-induced double-strand breaks and immune responses against AAV capsid proteins [123]. Strategies to mitigate these risks include using smaller Cas orthologs (SaCas9, CjCas9) and transient immunosuppression.
- Lentiviruses: Primarily used for ex vivo applications, offering stable genomic integration but with associated insertional mutagenesis risks.
Non-Viral Delivery Systems:
- Lipid Nanoparticles (LNPs): Emerging as a preferred delivery method for in vivo applications due to flexible cargo size, reduced immunogenicity, and potential for redosing [26]. Recent clinical data demonstrate that LNP-delivered CRISPR systems (e.g., Intellia's NTLA-2001 for hATTR) can be safely readministered, as LNPs don't trigger the same immune responses as viral vectors [26]. LNPs show natural tropism for liver tissue, making them particularly suitable for hepatocyte-targeted therapies.
- Electroporation: Mainly used for ex vivo delivery to hematopoietic stem cells and immune cells, with optimization required to maintain cell viability and editing efficiency.
Immunogenicity Assessment
CRISPR systems derived from bacterial proteins pose inherent immunogenicity risks. Comprehensive assessment includes:
- Pre-existing Immunity: Screening for pre-existing antibodies against Cas proteins in patient populations, as bacterial exposure may have primed immune responses.
- Cellular Immune Responses: Evaluation of T-cell responses against Cas epitopes, which could lead to clearance of edited cells.
- Mitigation Strategies: Approaches include using less immunogenic Cas orthologs, engineering Cas proteins to remove immunodominant epitopes, and transient immunosuppression during treatment.
Experimental Protocols for Safety Assessment
Comprehensive Off-Target Analysis Workflow
A robust off-target assessment protocol should integrate multiple complementary methods to provide comprehensive coverage:
Protocol: Integrated Off-Target Assessment
Step 1: In Silico Prediction
- Utilize multiple computational tools (e.g., Cas-OFFinder, COSMID) to identify potential off-target sites with up to 5 nucleotide mismatches and/or bulges.
- Include analysis of sequences with non-canonical PAM sites when relevant to the Cas variant being used.
- Prioritize sites in coding regions, regulatory elements, and known oncogenic/tumor suppressor loci.
Step 2: In Vitro Cleavage Assays
- Synthesize potential off-target sites identified in Step 1 as double-stranded DNA oligonucleotides.
- Incubate target sites with CRISPR ribonucleoprotein complexes under optimal reaction conditions.
- Measure cleavage efficiency using gel electrophoresis or fluorescence-based methods.
- Calculate specificity scores based on relative cleavage efficiency between on-target and off-target sites.
Step 3: Cell-Based Validation
- Transfer relevant cell types with CRISPR components using appropriate delivery methods.
- After 48-72 hours, harvest genomic DNA and prepare sequencing libraries for targeted amplicon sequencing of predicted off-target sites.
- Analyze sequencing data for insertion/deletion (indel) mutations at each potential off-target site using tools like CRISPResso2.
- Establish a threshold of significance (typically >0.1% indels with statistical significance) for reporting off-target activity.
Step 4: Unbiased Genome-Wide Screening
- Implement CIRCLE-Seq for in vitro, genome-wide off-target identification using purified genomic DNA.
- For in vivo contexts, utilize DISCOVER-Seq or related methods that exploit endogenous DNA repair markers.
- Sequence at sufficient depth (>50x coverage) to detect low-frequency events.
- Validate top hits from unbiased screens using targeted amplicon sequencing.
Step 5: Integrated Risk Assessment
- Compile results from all methods to create a comprehensive off-target profile.
- Prioritize off-target sites based on editing frequency, functional potential, and validation across multiple methods.
- For clinical applications, include a plan for long-term monitoring of top off-target sites in treated patients.
In Vivo Toxicology Study Design
Protocol: Comprehensive Toxicology Assessment for CRISPR Therapeutics
Study Objectives:
- Identify potential toxicities related to on-target editing in unintended tissues
- Assess consequences of off-target editing events
- Evaluate immune responses to CRISPR components or delivery vehicles
- Determine maximum tolerated dose and identify target organs of toxicity
Experimental Design:
- Species Selection: Use pharmacologically relevant species that demonstrate similar editing efficiency and distribution as expected in humans. For novel editors, include both rodent and non-rodent species when possible.
- Dosing Regimen: Include at least three dose levels spanning the anticipated clinical exposure range, plus a vehicle control group. Consider both single and multiple dosing schedules if relevant to clinical use.
- Group Size: Minimum of 10 animals per sex per group for main study cohorts, with additional animals included for recovery groups if assessing reversibility of effects.
- Study Duration: Typically 4-6 weeks for initial toxicity assessment, with longer-term studies (3-6 months) to evaluate persistence of editing and delayed effects.
Endpoint Assessments:
- Clinical Observations: Daily cage-side observations, detailed physical examinations weekly, body weight and food consumption measurements.
- Clinical Pathology: Comprehensive hematology, coagulation, and clinical chemistry panels at multiple timepoints post-dosing.
- Histopathology: Complete tissue collection and microscopic evaluation of all major organs, with special attention to delivery-relevant tissues (e.g., liver for LNP delivery).
- Editing Assessment: Quantitative measurement of on-target editing efficiency in target and potential off-target tissues. Assessment of persistence of editing over time.
- Immune Response Evaluation: Characterization of humoral and cellular immune responses against CRISPR components and delivery vehicles.
Statistical Analysis:
- Appropriate statistical methods for comparison to control groups (e.g., one-way ANOVA with post-hoc tests).
- Dose-response relationships should be assessed for key findings.
- Consideration of multiplicity in analysis of large parameter sets.
Safety Visualization Workflow
The following diagram illustrates the comprehensive safety assessment workflow for CRISPR therapeutics:
Emerging Challenges and Future Directions
Personalized CRISPR Therapeutics and Regulatory Innovation
The landmark case of a personalized CRISPR treatment for an infant with CPS1 deficiency demonstrates both the potential and challenges of bespoke gene editing therapies. Developed and delivered in just six months, this treatment required a multi-institutional collaboration and set a precedent for rapid regulatory approval of platform therapies [26] [126]. However, the scalability of this approach remains a significant challenge, as current manufacturing and regulatory frameworks are not designed for single-patient therapies.
The emerging regulatory framework for "platformization" of CRISPR therapies aims to address this challenge by creating streamlined pathways for therapies that share common elements. The FDA's proposal for approving on-demand gene editing medicines based on "plausible mechanism" and consistent patient benefit represents a potentially transformative approach to regulating personalized CRISPR therapeutics [125]. Academic institutions are pioneering this approach through umbrella trials for clinical syndromes like urea cycle disorders and severe T cell dysfunction, planned to begin in 2026 [125].
Manufacturing and Quality Control Challenges
The manufacturing requirements for CRISPR therapeutics present unique challenges, particularly for personalized approaches:
Quality Control Considerations:
- Potency Assays: Development of robust assays that accurately measure editing efficiency and functional outcomes.
- Purity Specifications: Establishing acceptable limits for process-related impurities and product-related variants.
- Characterization Methods: Comprehensive analysis of critical quality attributes including guide RNA integrity, Cas protein activity, and vector purity.
Manufacturing Innovation: Recent advances include "benefit-risk commensurate" accelerated small-scale manufacture developed through collaborations between academic institutions and industry partners [125]. These approaches aim to address the challenge of producing clinical-grade materials for patients with urgent medical needs where traditional manufacturing timelines are prohibitive.
Long-Term Monitoring and Post-Marketing Surveillance
As CRISPR therapies move toward broader clinical use, establishing frameworks for long-term safety monitoring becomes essential:
Key Elements of Long-Term Monitoring:
- Durability of Editing: Assessment of whether the therapeutic effect persists over time, particularly in dividing cell populations.
- Delayed Adverse Events: Monitoring for potential consequences that may manifest months or years after treatment.
- Germline Editing Exclusion: Confirmation that editing is restricted to somatic tissues and does not affect reproductive cells.
- Off-Target Surveillance: Ongoing assessment of potential long-term consequences of off-target editing events identified during development.
The regulatory and safety landscape for clinical CRISPR applications continues to evolve rapidly, with recent advances in platform-based regulation and safety assessment methodologies creating new pathways for therapeutic development. By integrating robust safety assessment protocols throughout the development process and engaging early with regulatory agencies, researchers can navigate this complex landscape while advancing innovative treatments for patients with serious genetic diseases.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) system represents a transformative advancement in genome editing technology. Originally identified as an adaptive immune system in bacteria and archaea, CRISPR-Cas9 has been adapted as a programmable tool for precise gene modification in eukaryotic cells [128] [3]. This RNA-guided system enables researchers to make targeted double-strand breaks (DSBs) in DNA, which are then repaired by the cell's endogenous repair mechanisms—predominantly non-homologous end joining (NHEJ) or homology-directed repair (HDR) [128] [3]. The simplicity, cost-effectiveness, and precision of CRISPR-Cas9 have positioned it as a promising therapeutic platform for addressing genetic disorders that traditional treatments cannot cure [128] [129].
This case study analysis examines real-world efficacy and safety data emerging from clinical trials of CRISPR-based therapies. Framed within the broader context of basic CRISPR research principles, we focus on the translation of these fundamental mechanisms into therapeutic applications across multiple disease areas, including hematologic disorders, genetic diseases, and oncology. The analysis incorporates quantitative efficacy metrics, safety profiles, detailed experimental methodologies, and visualization of key mechanistic and workflow concepts to provide a comprehensive technical resource for researchers, scientists, and drug development professionals.
Core Principles of CRISPR-Cas9 Gene Editing
Molecular Mechanisms and DNA Repair Pathways
The CRISPR-Cas9 system operates as a two-component complex consisting of the Cas9 nuclease and a guide RNA (gRNA) [128] [3]. The gRNA is a chimeric single guide RNA (sgRNA) that combines the functions of the endogenous CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA). The crRNA segment, containing a ~20 nucleotide spacer sequence, confers specificity through complementary base pairing with the target DNA sequence. This target sequence must be adjacent to a protospacer adjacent motif (PAM), typically 5'-NGG-3' for the commonly used Streptococcus pyogenes Cas9 (SpCas9) [128] [3].
Upon formation of the Cas9-gRNA complex and localization to the target DNA, the HNH and RuvC nuclease domains of Cas9 cleave the complementary and non-complementary DNA strands, respectively, generating a DSB [3]. The cellular repair of this break determines the editing outcome. The error-prone NHEJ pathway frequently results in small insertions or deletions (indels) that disrupt gene function, enabling gene knockout strategies. The less frequent HDR pathway uses a donor DNA template to facilitate precise nucleotide changes or gene insertions, but requires the presence of a repair template and occurs primarily in cycling cells [128] [3].
Figure 1: CRISPR-Cas9 Mechanism and DNA Repair Pathways. The Cas9-gRNA complex binds to target DNA adjacent to a PAM sequence, creating a double-strand break that is repaired via NHEJ (gene knockout) or HDR (precise editing).
Advanced CRISPR System Development
Beyond standard CRISPR-Cas9, several advanced editing platforms have been developed to address limitations in targeting range, precision, and safety. Base editing systems fuse a catalytically impaired Cas9 (nCas9) with a nucleobase deaminase enzyme, enabling direct chemical conversion of one DNA base to another without inducing DSBs. Cytosine base editors (CBEs) convert C•G to T•A base pairs, while adenine base editors (ABEs) convert A•T to G•C base pairs [128] [3]. Prime editing utilizes a Cas9-reverse transcriptase fusion protein and a prime editing guide RNA (pegRNA) to directly write new genetic information into a target DNA site, enabling all 12 possible base-to-base conversions as well as small insertions and deletions without DSBs [128]. Additionally, CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) employ catalytically dead Cas9 (dCas9) fused to repressor or activator domains to modulate gene transcription without altering the underlying DNA sequence [130].
Clinical Trial Methodologies and Experimental Protocols
Clinical Grade CRISPR Therapeutic Development
The translation of CRISPR systems from research tools to clinical therapeutics requires rigorous manufacturing protocols and quality control measures. Clinical trials follow standardized phases (I-III) to evaluate safety, dosage, efficacy, and therapeutic benefit [26]. The methodologies below detail key approaches for major CRISPR therapeutic platforms.
Ex Vivo Hematopoietic Stem Cell (HSC) Editing Protocol
The methodology for CASGEVY (exagamglogene autotemcel), the first FDA-approved CRISPR-based therapy for sickle cell disease (SCD) and transfusion-dependent beta thalassemia (TDT), exemplifies the ex vivo editing approach [26] [131]:
HSC Collection: CD34+ hematopoietic stem and progenitor cells are collected from the patient via apheresis after mobilization from bone marrow to peripheral blood using granulocyte colony-stimulating factor (G-CSF).
CRISPR-Cas9 Electroporation:
- Cells are transfected with CRISPR-Cas9 components targeting the BCL11A erythroid-specific enhancer region via electroporation.
- The editing machinery is typically delivered as ribonucleoprotein (RNP) complexes to minimize off-target effects and reduce immunogenicity.
Myeloablative Conditioning: Patients undergo busulfan conditioning to create marrow niche space for the edited cells.
Reinfusion: The CRISPR-edited CD34+ cells are infused back into the patient, where they engraft in the bone marrow and reconstitute hematopoiesis.
Monitoring and Validation:
- Engraftment is monitored through absolute neutrophil and platelet counts.
- Editing efficacy is assessed by measuring fetal hemoglobin (HbF) levels using high-performance liquid chromatography.
- BCL11A gene modification is confirmed via next-generation sequencing of colony-forming unit assays.
In Vivo Lipid Nanoparticle (LNP) Delivery Protocol
For direct in vivo administration, as demonstrated in trials for hereditary transthyretin amyloidosis (hATTR) and hereditary angioedema (HAE), CRISPR components are delivered systemically using lipid nanoparticles (LNPs) [26]:
Formulation: CRISPR-Cas9 mRNA and sgRNA are encapsulated in LNPs optimized for hepatocyte tropism. The LNP formulation typically comprises ionizable lipids, phospholipids, cholesterol, and PEGylated lipids.
Dosing Administration: LNPs are administered via intravenous infusion at dose levels determined from preclinical studies. Dose escalation follows a modified 3+3 design in Phase I trials.
Pharmacodynamic Assessment:
- For hATTR: Serum transthyretin (TTR) protein levels are quantified as a biomarker of editing efficacy.
- For HAE: Plasma kallikrein levels and the frequency of HAE attacks are monitored.
Safety Monitoring: Comprehensive assessment includes monitoring for infusion-related reactions, liver function tests (ALT, AST), anti-Cas9 antibodies, and potential off-target effects through unbiased genome-wide assays.
Allogeneic CAR-T Cell Manufacturing Protocol
For allogeneic CAR-T therapies (e.g., CTX112 for B-cell malignancies), the process involves multiplexed editing to generate universal, off-the-shelf cell products [131]:
T Cell Isolation: T cells are collected from healthy donors via leukapheresis.
Multiplex CRISPR Editing:
- Cells are electroporated with RNP complexes targeting multiple loci: TRAC (T cell receptor alpha constant) to reduce graft-versus-host disease, CD52 to confer alemtuzumab resistance, and a CD19-CAR insertion via HDR using an AAV6 donor template.
- Editing efficiency is validated via flow cytometry and DNA sequencing.
Expansion and Formulation: Edited T cells are expanded ex vivo using cytokine stimulation (IL-2, IL-7, IL-15), then cryopreserved in infusion-ready vials.
Lymphodepletion and Administration: Patients receive lymphodepleting chemotherapy (fludarabine/cyclophosphamide) followed by infusion of allogeneic CAR-T cells.
Response Assessment: Tumor response is evaluated using Lugano criteria, while cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome are graded using ASTCT criteria.
Figure 2: CRISPR Therapeutic Workflows. Three primary approaches for CRISPR-based therapies: ex vivo editing of patient cells, in vivo systemic delivery via LNPs, and multiplexed editing of allogeneic donor cells for universal therapies.
Case Studies: Efficacy and Safety Data Analysis
Quantitative Efficacy and Safety Data from Clinical Trials
Table 1: Clinical Efficacy and Safety Data from Selected CRISPR Trials
| Therapeutic (Condition) | Target/Mechanism | Trial Phase | Key Efficacy Metrics | Safety Profile |
|---|---|---|---|---|
| CASGEVY (exa-cel) (SCD/TDT) [26] [131] | BCL11A erythroid enhancer (HbF induction) | Approved (Phase III) | SCD: 94.5% freedom from vaso-occlusive crises (≥12 months) [26].TDT: 92.7% transfusion independence (≥12 months) [26]. | Myeloablative conditioning toxicity; manageable adverse events consistent with autologous transplant. No off-target concerns identified in comprehensive assessments. |
| NTLA-2001 (hATTR) [26] | TTR gene knockout (LNP delivery) | Phase III | ~90% sustained reduction in serum transthyretin (TTR) at 2 years (27/27 patients) [26]. Disease stabilization/improvement in neuropathy/cardiomyopathy symptoms. | Mild to moderate infusion-related reactions; no serious treatment-related adverse events; no liver safety signals identified. |
| NTLA-2002 (HAE) [26] | KLKB1 gene knockout (kallikrein inhibition) | Phase I/II | 86% reduction in plasma kallikrein; 88% attack-free (16 weeks post-treatment) in high-dose group [26]. | Favorable safety profile; no dose-limiting toxicities observed. |
| CTX112 (B-cell malignancies) [131] | Allogeneic anti-CD19 CAR-T | Phase I/II | 100% response rate (6/6 patients) in prior T-cell engager failures; robust CAR-T expansion [131]. | Manageable CRS; no ICANS; no GvHD despite allogeneic source. RMAT designation granted. |
Comprehensive Safety Analysis Across Trials
The safety profile of CRISPR-based therapies has been systematically evaluated across clinical trials, with several consistent observations:
On-Target Safety: The most significant safety considerations relate to the intended biological effects rather than off-target editing. For CASGEVY, the primary risks are associated with myeloablative conditioning (busulfan) including cytopenias, infections, and secondary malignancies [26] [131]. For in vivo LNP-delivered therapies, infusion-related reactions constitute the most frequently reported adverse events [26].
Off-Target Editing Analysis: Comprehensive genomic assessment across multiple trials has not identified clinically significant off-target effects. In CASGEVY trials, unbiased genome-wide sequencing approaches detected no off-target editing above background mutation rates [26]. Similarly, Intellia's in vivo programs have reported clean safety profiles without evidence of treatment-related genotoxicity [26].
Immunogenicity: Immune responses against bacterial-derived Cas9 protein represent a theoretical concern. However, in LNP-delivered therapies where redosing is possible, the absence of severe immune reactions against Cas9 has been demonstrated. The LNP delivery system appears less immunogenic than viral vectors, enabling potential redosing as evidenced by the hATTR trial where participants received second infusions [26].
Long-Term Safety: Durability of response and long-term safety continue to be monitored. For CASGEVY, sustained efficacy beyond 2 years with stable HbF levels has been observed without late-onset toxicities [26]. In the hATTR trial, 27 participants maintained ~90% TTR reduction through 2 years of follow-up with no evidence of diminishing effect or late adverse events [26].
Research Reagent Solutions and Technical Tools
Table 2: Essential Research Reagents and Platforms for CRISPR Clinical Translation
| Reagent/Platform | Function | Clinical Application Example |
|---|---|---|
| Lipid Nanoparticles (LNPs) | In vivo delivery of CRISPR components; hepatotropic | Intellia's hATTR and HAE programs [26] |
| Ribonucleoprotein (RNP) Complexes | Direct delivery of preassembled Cas9-gRNA; reduced off-target effects | CASGEVY HSC editing [26] [131] |
| AAV Donor Templates | High-efficiency HDR mediation for precise gene insertion | CAR-T cell engineering (CTX112) [131] |
| Guide RNA Libraries | Genome-wide screening for target identification | Functional genomics in drug discovery [130] [132] |
| Next-Generation Sequencing (NGS) | Off-target assessment; editing efficiency quantification | Regulatory safety packages [26] [132] |
| Base Editors (BE) | Single-nucleotide conversion without DSBs | Precision correction of point mutations in ongoing trials [128] [3] |
| Prime Editors (PE) | Versatile editing (all base changes, small indels) without DSBs | Preclinical development for expanded therapeutic applications [128] |
The accumulation of real-world clinical trial data demonstrates that CRISPR-based therapies can deliver transformative efficacy for genetic diseases with generally manageable safety profiles. The approval of CASGEVY and the advanced clinical progress of multiple programs validate the therapeutic application of CRISPR technology. The consistent observation of durable clinical benefits without significant off-target effects across independent trials provides reassurance about the specificity of CRISPR-based gene editing in humans.
Future directions in CRISPR therapeutics include: (1) expanding the range of addressable diseases through improved delivery systems, particularly LNPs with tropism for organs beyond the liver; (2) enhancing precision through next-generation editors like base and prime editors; (3) reducing manufacturing complexity and cost through in vivo approaches; and (4) developing personalized therapies for ultra-rare diseases, as demonstrated by the bespoke CRISPR treatment for CPS1 deficiency developed within six months [26]. As the field advances, addressing challenges related to manufacturing scalability, equitable access, and long-term monitoring will be essential for realizing the full potential of CRISPR-based medicines across broader patient populations.
Conclusion
CRISPR technology has fundamentally transformed biomedical research and therapeutic development by providing an unprecedented ability to precisely manipulate the genome. Its simplicity, versatility, and cost-effectiveness have made it the dominant gene-editing platform, surpassing older technologies like ZFNs and TALENs. While challenges such as off-target effects, efficient delivery, and editing certain genomic regions remain, ongoing innovations in high-fidelity enzymes, novel delivery systems, and advanced editing techniques like base and prime editing are rapidly addressing these limitations. For researchers and drug developers, a deep understanding of both foundational principles and advanced optimization strategies is crucial for successfully applying CRISPR. The future of CRISPR lies in refining its precision and safety for broader clinical translation, expanding its use in multiplexed editing and complex disease modeling, and integrating it with other modalities to realize the full potential of precision medicine across a wide spectrum of genetic disorders, cancers, and neurodegenerative diseases.
References
- 1. CRISPR-Cas Systems: Bridging Bacterial Immunity and ... [https://www.mdpi.com/2673-8007/5/4/118]
- 2. How Bacteria Use CRISPR to Vaccinate against Viruses [https://www.scientificamerican.com/article/how-bacteria-use-crispr-to-vaccinate-against-viruses/]
- 3. Advancing CRISPR genome editing into gene therapy clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12094669/]
- 4. Advancements in CRISPR/Cas systems for disease treatment [https://pmc.ncbi.nlm.nih.gov/articles/PMC12254762/]
- 5. An updated evolutionary classification of CRISPR–Cas ... [https://www.nature.com/articles/s41564-025-02180-8]
- 6. Screening approach enhances CRISPR genome-editing ... [https://www.stjude.org/media-resources/news-releases/2025-medicine-science-news/screening-approach-enhances-crispr-genome-editing-efficiency.html]
- 7. Crystal Structure of Cas9 in Complex with Guide RNA and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4139937/]
- 8. CRISPR Guide [https://www.addgene.org/guides/crispr/]
- 9. The Complete Guide to Understanding CRISPR sgRNA [https://www.synthego.com/guide/how-to-use-crispr/sgrna/]
- 10. characteristics of functional guide RNAs for the CRISPR/Cas9 ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0784-0]
- 11. Internal guide RNA interactions interfere with Cas9- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4906408/]
- 12. CRISPR-Cas9 and Its Bioinformatics Tools: A Systematic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12109910/]
- 13. CRISPR-induced double-strand breaks trigger ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6587125/]
- 14. Qualitative and Quantitative Detection of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10572612/]
- 15. Using the safety scissors: DNA resection regulation at DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599495/]
- 16. Beyond the Double-Stranded Cut: Evolution of CRISPR ... [https://www.synthego.com/blog/beyond-crispr-methods/]
- 17. A CRISPR Interference Screen of Essential Genes Reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8407339/]
- 18. How to Pick the Best CRISPR Data Analysis Method for ... [https://www.synthego.com/guide/how-to-use-crispr/analysis-methods/]
- 19. A Scaled Framework for CRISPR Editing of Human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5639480/]
- 20. HDR vs NHEJ: DNA Repair Pathway Comparison [https://invivobiosystems.com/crispr/hdr-vs-nhej/]
- 21. HDR vs. NHEJ: CRISPR Gene Editing Techniques [https://www.zeclinics.com/blog/hdr-vs-nhej-in-crispr-gene]
- 22. CRISPR 101: Homology Directed Repair [https://blog.addgene.org/crispr-101-homology-directed-repair]
- 23. CRISPR-Cas9-mediated homology-directed repair for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11531618/]
- 24. NHEJ vs HDR: Difference when Repairing CRISPR DSB [https://www.idtdna.com/pages/support/faqs/which-repair-pathway-is-most-commonly-used-to-repair-crispr-mediated-double-stranded-breaks-nhej-or-hdr]
- 25. DNA repair pathway choices in CRISPR-Cas9 mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8187289/]
- 26. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 27. CMN Weekly (12 September 2025) [https://crisprmedicinenews.com/news/cmn-weekly-12-september-2025-your-weekly-crispr-medicine-news/]
- 28. [Latest] Global CRISPR Gene Editing Market Size/ ... [https://finance.yahoo.com/news/latest-global-crispr-gene-editing-193000031.html]
- 29. AI-powered CRISPR could lead to faster gene therapies ... [https://med.stanford.edu/news/all-news/2025/09/ai-crispr-gene-therapy.html]
- 30. The next generation of CRISPR–Cas technologies and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7079207/]
- 31. A Review on the Mechanism and Applications of CRISPR/ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9512960/]
- 32. Structures, mechanisms and applications of RNA ... - PMC - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC11375968/]
- 33. CRISPR-Cas12 and Cas13: the lesser known siblings of ... [https://link.springer.com/article/10.1007/s10565-019-09489-1]
- 34. Assessing and advancing the safety of CRISPR-Cas tools [https://www.nature.com/articles/s41467-023-35886-6]
- 35. CRISPR-Cas12/Cas13: Bibliometric analysis and ... [https://www.sciencedirect.com/science/article/pii/S1876034124000534]
- 36. CRISPR/Cas9‐mediated genome editing [https://pmc.ncbi.nlm.nih.gov/articles/PMC7171402/]
- 37. A Guide to Efficient CRISPR gRNA Design: Principles and ... [https://www.genscript.com/a-guide-to-efficient-crispr-grna-design-principles-and-design-tools.html]
- 38. CRISPR: Guide to gRNA design [https://www.snapgene.com/guides/design-grna-for-crispr]
- 39. How To Design Guide RNA for CRISPR [https://www.synthego.com/guide/how-to-use-crispr/design-grna-crispr/]
- 40. CRISPR guide RNA design for research applications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5014588/]
- 41. crispr [https://genome.ucsc.edu/training/education/crispr.html]
- 42. Research Techniques Made Simple: CRISPR Genetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8525197/]
- 43. New software promises to make precision genome editing ... [https://news.ucsc.edu/2025/07/crisprware/]
- 44. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 45. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 46. Challenges and Opportunities in the Application of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503735/]
- 47. 5 Reasons Why Ribonucleoproteins Are a Better ... [https://www.synthego.com/blog/crispr-plasmid-pitfalls/]
- 48. innovations and challenges in rAAV vector-based CRISPR ... [https://www.nature.com/articles/s41434-025-00573-2]
- 49. Viral Vector vs CRISPR Resources [https://bioinnovatise.com/articles/viral-vector-vs-crispr/]
- 50. Overcoming the Delivery Challenges in CRISPR/Cas9 ... [https://www.medsci.org/v22p3625.htm]
- 51. CRISPR Cell and Gene Therapies: Key Challenges in ... [https://www.synthego.com/blog/crispr-cell-gene-therapy-obstacles/]
- 52. What is CRISPR? A bioengineer explains - Stanford Report [https://news.stanford.edu/stories/2024/06/stanford-explainer-crispr-gene-editing-and-beyond]
- 53. Annotation and Classification of CRISPR-Cas Systems - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5901762/]
- 54. Perturbomics: CRISPR–Cas screening-based functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322284/]
- 55. CRISPR Screens: Approaches, Strategies, and Workflow [https://www.synthego.com/guide/crispr-methods/crispr-screen/]
- 56. Large-scale CRISPR screening in primary human 3D ... [https://www.nature.com/articles/s41467-025-62818-3]
- 57. Recent advances in therapeutic gene-editing technologies [https://www.sciencedirect.com/science/article/pii/S152500162500200X]
- 58. How To Use ICE: A Detailed Guide for Analyzing CRISPR ... [https://www.synthego.com/guide/how-to-use-crispr/ice-analysis-guide/]
- 59. Sickle Cell Gene Therapy Using CRISPR [https://www.synthego.com/crispr-sickle-cell-disease/]
- 60. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β- ... [https://pubmed.ncbi.nlm.nih.gov/33283989/]
- 61. Gene Therapy May Cure Sickle Cell Disease and Thalassemia [https://consultqd.clevelandclinic.org/gene-therapy-trials-show-positive-results-in-sickle-cell-disease-and-thalassemia]
- 62. Characterizing and controlling CRISPR repair outcomes in ... [https://www.nature.com/articles/s41467-025-66058-3]
- 63. Emerging trends in prime editing for precision genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322275/]
- 64. Current Status and Challenges of DNA Base Editing Tools [https://pmc.ncbi.nlm.nih.gov/articles/PMC7474268/]
- 65. A Review on Advanced CRISPR - Based - Genome Tools: Editing ... Base [https://link.springer.com/article/10.1007/s12033-022-00639-1]
- 66. From bench to bedside: cutting-edge applications of base and... editing [https://pmc.ncbi.nlm.nih.gov/articles/PMC11662623/]
- 67. CRISPR Base Editing Explained | ABE & CBE in Gene ... [https://www.synthego.com/crispr-base-editing-guide/]
- 68. Prime editing: therapeutic advances and mechanistic insights [https://www.nature.com/articles/s41434-024-00499-1]
- 69. Prime editing: Mechanism insight and recent applications in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10754014/]
- 70. Single prime editing system could potentially treat multiple ... [https://www.broadinstitute.org/news/single-prime-editing-system-could-potentially-treat-multiple-genetic-diseases]
- 71. Personalized gene editing helped one baby: can it be ... [https://www.nature.com/articles/d41586-025-03566-8]
- 72. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 73. The hidden risks of CRISPR/Cas: structural variations and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12325979/]
- 74. High-fidelity Cas9-mediated targeting of KRAS driver ... [https://www.nature.com/articles/s41467-025-62350-4]
- 75. CMN Weekly (31 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-31-october-2025-your-weekly-crispr-medicine-news/]
- 76. Comparative Analysis of Methods for Assessing On-Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11932317/]
- 77. Heterochromatin delays CRISPR-Cas9 mutagenesis but does ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6306241/]
- 78. Strategies to overcome the main challenges of the use of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8892709/]
- 79. Why Are Some Genes Difficult to CRISPR Edit? [https://www.synthego.com/blog/genes-difficult-crispr-edit/]
- 80. CRISPRa and CRISPRi [https://www.synthego.com/guide/crispr-methods/crispri-crispra/]
- 81. The therapeutic promises of CRISPRa and CRISPRi [https://www.sciencedirect.com/science/article/pii/S1525001623001454]
- 82. Cornerstones of CRISPR-Cas in drug development and therapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC5459481/]
- 83. CRISPRi and CRISPRa screens in mammalian cells for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5886776/]
- 84. CRISPRa and CRISPRi: Why You Need These Powerful ... [https://bitesizebio.com/47575/crispra-and-crispri/]
- 85. A versatile, high-efficiency platform for CRISPR-based ... [https://www.nature.com/articles/s41467-023-36452-w]
- 86. Novel drug-inducible CRISPRa/i systems for rapid and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12185803/]
- 87. Comparative CRISPRi screens reveal a human stem cell ... [https://www.nature.com/articles/s41594-025-01616-3]
- 88. The Potential and Pitfalls of CRISPRa [https://frontlinegenomics.com/crispra-therapeutic-applications/]
- 89. Genomic copy number dictates a gene-independent cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4972686/]
- 90. Applications of multiplexed CRISPR–Cas for genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322214/]
- 91. New CRISPR/Cas9-based system models NAHR copy- ... [https://www.jax.org/news-and-insights/2016/february/crispr-system-models-nahr-cnvs]
- 92. CMN Weekly (26 September 2025) [https://crisprmedicinenews.com/news/cmn-weekly-26-september-2025-your-weekly-crispr-medicine-news/]
- 93. GUIDE-Seq enables genome-wide profiling of off-target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4320685/]
- 94. CRISPR Technology in Disease Management [https://pmc.ncbi.nlm.nih.gov/articles/PMC12584872/]
- 95. CIRCLE-seq: a highly sensitive in vitro screen for genome- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5924695/]
- 96. CIRCLE-Seq for Interrogation of Off-Target Gene Editing [https://pubmed.ncbi.nlm.nih.gov/39555799/]
- 97. Genome-wide detection of CRISPR editing in vivo using ... [https://www.nature.com/articles/s41467-022-28135-9]
- 98. TEG-Seq CRISPR Off-Target Analysis [https://www.thermofisher.com/us/en/home/products-and-services/services/custom-services/teg-seq-crispr-off-target-analysis.html]
- 99. Massively parallel CRISPR off-target detection enables ... [https://www.sciencedirect.com/science/article/pii/S2666634023001630]
- 100. Guidelines for Performing CRISPR/Cas9 Genome Editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10610170/]
- 101. Sanger Sequencing vs. Next-Generation Sequencing (NGS) [https://www.genscript.com/gene-news/sanger-sequencing-vs-next-generation-sequencing.html]
- 102. Next-Generation Sequencers vs. Sanger ... [https://www.labx.com/resources/next-generation-sequencers-vs-sanger-sequencers-key-differences/5597]
- 103. Sanger or NGS DNA Sequencing: Which is the Best ... [https://www.biobasic.com/blog/sanger-or-ngs-dna-sequencing-which-is-the-best-technology.html]
- 104. NGS vs Sanger Sequencing [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/advantages/ngs-vs-sanger.html]
- 105. Next-generation sequencing: 2025 Revolution [https://lifebit.ai/blog/next-generation-sequencing/]
- 106. High-Throughput Genotyping | CRISPR Validation [https://www.genewiz.com/public/services/next-generation-sequencing/high-throughput-genotyping]
- 107. The Future of Sanger Sequencing: Technical Development ... [https://www.cd-genomics.com/blog/sanger-sequencing-technology-innovation/]
- 108. Sanger sequencing is no longer always necessary based ... [https://www.nature.com/articles/s41598-021-85182-w]
- 109. Systematic Evaluation of Sanger Validation of NextGen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4878677/]
- 110. Comparing Gene Editing Platforms: CRISPR vs. Traditional ... [https://blog.crownbio.com/comparing-gene-editing-platforms-crispr-vs-traditional-methods]
- 111. A Comparative Review of Genetic Modification Tools for ... [https://nhsjs.com/2025/a-comparative-review-of-genetic-modification-tools-for-crop-improvement-efficiency-scalability-and-usability/]
- 112. State of CRISPR Gene Editing in the Drug Discovery Sector [https://www.synthego.com/blog/survey-report-crispr-therapeutics/]
- 113. CRISPR-based Gene Editing Market Size to Reach USD ... [https://www.biospace.com/press-releases/crispr-based-gene-editing-market-size-to-reach-usd-24-37-billion-by-2034]
- 114. Gene Editing Market Trends, Share and Forecast, 2025-2032 [https://www.coherentmarketinsights.com/industry-reports/gene-editing-market]
- 115. CRISPR Market Size Hits USD 3.27 Billion in 2025 [https://www.towardshealthcare.com/insights/crispr-market-sizing]
- 116. Overview CRISPR Clinical Trials 2025 - Learn | Innovate [https://crisprmedicinenews.com/clinical-trials/]
- 117. Genome Editing Market Trends, Share and Analysis, 2025- ... [https://www.coherentmi.com/industry-reports/genome-editing-market]
- 118. Overcoming the Delivery Challenges in CRISPR/Cas9 Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12434829/]
- 119. Scientists just made CRISPR three times more effective [https://www.sciencedaily.com/releases/2025/09/250907024543.htm]
- 120. CRISPR This Way: Choosing Between Delivery System [https://en.vectorbuilder.com/resources/vector-academy/lessons/crispr-delivery-systems.html]
- 121. Off-target effects in CRISPR-Cas genome editing for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12329535/]
- 122. Ensure Proper Controls in Your CRISPR Experiments [https://www.synthego.com/blog/controls-crispr-experiments/]
- 123. Basic Principles and Clinical Applications of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8819410/]
- 124. CRISPR Therapeutics Provides First Quarter 2025… [https://crisprtx.com/about-us/press-releases-and-presentations/crispr-therapeutics-provides-first-quarter-2025-financial-results-and-announces-positive-top-line-data-from-phase-1-clinical-trial-of-ctx310-targeting-angptl3]
- 125. Celebrating a new, faster path to gene-editing medicines on demand [https://www.statnews.com/2025/11/14/fda-gene-editing-medicines-on-demand-pathway/]
- 126. Progress and Pressure: Taking Stock of the CRISPR Landscape [https://www.medscape.com/viewarticle/progress-and-pressure-taking-stock-crispr-landscape-2025a1000rzr]
- 127. CRISPR Clinical Trials to Follow [https://www.synthego.com/blog/crispr-clinical-trials/]
- 128. Current trends of clinical trials involving CRISPR/Cas ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10666174/]
- 129. Advancements in CRISPR/Cas systems for disease treatment [https://www.sciencedirect.com/science/article/pii/S2211383525003132]
- 130. CRISPR: A Screener's Guide - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7036479/]
- 131. Therapeutics Highlights Strategic Priorities and... - BioSpace CRISPR [https://www.biospace.com/press-releases/crispr-therapeutics-highlights-strategic-priorities-and-anticipated-2025-milestones]
- 132. High-content CRISPR screening [https://www.nature.com/articles/s43586-021-00093-4]