Bridging Accuracy and Efficiency: Integrating DFT and Machine Learning for Next-Generation Material Validation
This article explores the transformative integration of Density Functional Theory (DFT) and Machine Learning (ML) for validating material properties, a critical step in accelerating materials discovery and drug development.
Bridging Accuracy and Efficiency: Integrating DFT and Machine Learning for Next-Generation Material Validation
Abstract
This article explores the transformative integration of Density Functional Theory (DFT) and Machine Learning (ML) for validating material properties, a critical step in accelerating materials discovery and drug development. We first establish the foundational synergy between DFT's accuracy and ML's scalability. The piece then delves into cutting-edge methodological frameworks, from specialized force fields to modular learning, and provides crucial troubleshooting strategies for overcoming data scarcity and generalization challenges. Finally, we present a rigorous comparative analysis of ML model performance and validation protocols, offering researchers and scientists a comprehensive guide to confidently deploying these hybrid computational approaches for reliable material property prediction.
The Computational Synergy: How DFT and Machine Learning Are Revolutionizing Materials Science
Density Functional Theory (DFT) stands as a cornerstone of modern computational materials science, enabling the prediction of electronic structures and material properties from first principles. Despite its widespread adoption, DFT is fundamentally constrained by its high computational cost, which typically scales as the cube of the system size (~O(N³)). This cubic scaling limits routine calculations to systems comprising only a few hundred atoms, creating a significant bottleneck for researching experimentally relevant systems that often involve hundreds of thousands of atoms or more [1]. The core of this computational challenge lies in the solution of the Kohn-Sham equations, which requires iterative diagonalization of large Hamiltonian matrices—a process whose cost grows prohibitively with system size [2] [3].
Recent advancements in machine learning (ML) are now circumventing this long-standing limitation, enabling electronic structure predictions at unprecedented scales. By developing ML surrogates that emulate key aspects of DFT calculations, researchers have demonstrated up to three orders of magnitude speedup on systems where DFT is tractable and, more importantly, have enabled predictions on scales where DFT calculations are fundamentally infeasible [1]. This article examines the computational bottlenecks of traditional DFT and presents detailed protocols for implementing machine learning approaches that maintain quantum accuracy while achieving linear scaling.
Quantitative Analysis of DFT Bottlenecks and ML Solutions
Table 1: Computational Scaling and Performance Comparison of Traditional DFT vs. Machine Learning Approaches
| Method | Computational Scaling | Maximum Practical System Size | Speedup Factor | Key Limitations |
|---|---|---|---|---|
| Traditional DFT | O(N³) | Few hundred atoms | Reference | Cubic scaling from matrix diagonalization [1] |
| Linear-Scaling DFT | O(N) to O(N²) | Thousands of atoms | 10-100x | Limited generality, implementation complexity [1] |
| MALA (ML-LDOS) | O(N) | 100,000+ atoms | Up to 1000x | Training data requirements, transferability [1] [4] |
| ML-DFT Framework | O(N) with small prefactor | 10,000+ atoms | Orders of magnitude | Chemical space coverage [3] |
| SPHNet | O(N) with reduced TP operations | Extended molecular systems | 7x faster than prior ML models | Basis set limitations [2] |
Table 2: Accuracy Benchmarks of ML-DFT Methods Across Material Systems
| Method | Material System | Property Predicted | Error Metric | Performance |
|---|---|---|---|---|
| MALA | Beryllium with stacking faults | Formation energy | N⁻¹/³ scaling | Correct physical behavior [1] |
| ML-DFT Framework | Organic molecules (C, H, N, O) | Total energy | Chemical accuracy | ~1 kcal/mol [3] |
| ML-DFT Framework | Polymer chains & crystals | Atomic forces | MAE | Suitable for MD simulations [3] |
| Neural Network Correction | Al-Ni-Pd, Al-Ni-Ti alloys | Formation enthalpy | Improved agreement with experiment | Enhanced phase stability prediction [5] |
| DNN Model | Battery cathode materials | Average voltage | MAE | ~0.3-0.4 V vs DFT [6] |
Machine Learning Approaches to DFT Acceleration
Key Methodological Frameworks
3.1.1 Local Density of States Learning (MALA) The Materials Learning Algorithms (MALA) package addresses DFT scalability by training neural networks to predict the local density of states (LDOS) directly from atomic environments. This approach leverages the "nearsightedness" principle of electronic matter, which states that local electronic properties depend primarily on nearby atomic arrangements. MALA employs bispectrum coefficients as descriptors that encode the positions of atoms relative to every point in real space, enabling a feed-forward neural network to map these descriptors to the LDOS [1] [4]. Since this mapping is performed individually for each point in real space, the resulting workflow is highly parallelizable and scales linearly with system size.
3.1.2 Direct Hamiltonian Prediction (SPHNet) SPHNet represents an alternative approach that focuses on directly predicting the Hamiltonian matrix using SE(3)-equivariant graph neural networks. This method incorporates adaptive sparsity through two innovative gates: the Sparse Pair Gate filters out unimportant node pairs to reduce tensor product computations, while the Sparse TP Gate prunes less significant interactions across different orders in tensor products. A Three-phase Sparsity Scheduler ensures stable convergence, allowing SPHNet to achieve up to 70% sparsity while maintaining accuracy, resulting in a 7x speedup over previous models and reduced memory usage by up to 75% [2].
3.1.3 Charge Density Emulation A third paradigm involves end-to-end ML models that emulate the essence of DFT by mapping atomic structures directly to electronic charge densities, from which other properties are derived. These frameworks use atom-centered fingerprints to represent chemical environments and predict charge density descriptors, which then serve as inputs for predicting additional electronic and atomic properties. This strategy maintains consistency with the fundamental DFT principle that the electronic charge density determines all system properties [3].
Experimental Protocols
Protocol 1: ML-DFT Workflow for Large-Scale Electronic Structure Prediction
Figure 1: ML-DFT workflow for electronic structure calculation
Training Data Generation
- Perform DFT calculations on diverse small systems (typically 100-500 atoms) using standard packages (Quantum ESPRESSO, VASP).
- Calculate and store the local density of states (LDOS) or charge density on real-space grids.
- Extract atomic positions and convert to descriptors (bispectrum coefficients) using LAMMPS integration.
Model Training
- Implement feed-forward neural networks using PyTorch or TensorFlow.
- Train models to map bispectrum descriptors to LDOS values.
- Validate against held-out DFT calculations to ensure transferability.
Inference on Large Systems
- Deploy trained model to predict LDOS for large atomic configurations (>100,000 atoms).
- Post-process LDOS to compute electronic density, density of states, and total energy.
- Perform molecular dynamics simulations using predicted forces.
This protocol has demonstrated accurate energy calculations for beryllium systems with 131,072 atoms in just 48 minutes on 150 standard CPUs—orders of magnitude faster than conventional DFT [1] [4].
Protocol 2: Hamiltonian Prediction for Molecular Systems
Figure 2: SPHNet workflow for Hamiltonian prediction
Data Preparation
- Generate Hamiltonian matrices for diverse molecular configurations using DFT.
- Utilize datasets such as QH9 or PubchemQH containing quantum chemical properties.
Model Implementation
- Construct SE(3)-equivariant graph neural network with tensor product operations.
- Implement two sparse gates: Sparse Pair Gate for node pairs and Sparse TP Gate for tensor product interactions.
- Apply Three-phase Sparsity Scheduler (random → adaptive → fixed) to gradually increase sparsity to 70%.
Training and Validation
- Train model to predict Hamiltonian matrices from atomic structures.
- Evaluate on test molecules for accuracy in derived properties (energy, HOMO-LUMO gap).
- Benchmark computational performance against traditional DFT and other ML models.
This approach has demonstrated state-of-the-art accuracy on QH9 and PubchemQH datasets while providing up to 7x speedup over existing models [2].
Table 3: Key Software Packages and Datasets for ML-DFT Research
| Resource | Type | Primary Function | Application Scope |
|---|---|---|---|
| MALA | Software package | ML-driven electronic structure prediction | Large-scale materials simulation [1] [4] |
| SPHNet | Efficient neural network | Hamiltonian prediction with adaptive sparsity | Molecular systems with large basis sets [2] |
| Quantum ESPRESSO | DFT code | Generate training data & benchmarks | General electronic structure calculations [4] |
| LAMMPS | Molecular dynamics | Descriptor calculation & dynamics | Atomic-scale simulation [1] [4] |
| OMol25 | Dataset | 100M+ DFT molecular snapshots | Training generalizable ML potentials [7] [8] |
| Materials Project | Database | DFT-calculated material properties | Battery materials validation [6] |
Application Notes
Stacking Fault Energetics in Beryllium
The application of MALA to beryllium systems with stacking faults demonstrates the capability of ML-DFT to capture subtle energetic differences in extended defects. By introducing a stacking fault (shifting three atomic layers to change local crystal structure from hcp to fcc), researchers used ML predictions to compute the energetic differences between faulted and pristine systems. The results correctly followed the expected ~N⁻¹/³ scaling with system size, validating that ML-derived energies exhibit correct physical behavior even for systems of 131,072 atoms—far beyond conventional DFT capabilities [1].
Battery Voltage Prediction
Machine learning models trained on DFT data from the Materials Project have successfully predicted average voltages for alkali-metal-ion battery materials. Using deep neural networks with comprehensive feature sets (structural, physical, chemical, electronic, thermodynamic, and battery descriptors), researchers achieved close alignment with DFT calculations (MAE ~0.3-0.4 V). This approach enabled rapid screening of novel Na-ion battery compositions, with subsequent DFT validation confirming predicted voltages, demonstrating a viable hybrid ML-DFT workflow for accelerated materials discovery [6].
Alloy Phase Stability
For ternary alloy systems (Al-Ni-Pd and Al-Ni-Ti) relevant to high-temperature applications, neural network models have been employed to correct systematic errors in DFT-calculated formation enthalpies. By learning the discrepancy between DFT and experimental values using elemental concentrations, atomic numbers, and interaction terms as features, ML corrections significantly improved the accuracy of phase stability predictions, enabling more reliable determination of ternary phase diagrams [5].
The integration of machine learning with density functional theory represents a paradigm shift in computational materials research, effectively addressing the fundamental scalability limitations of traditional DFT. Through approaches ranging from local density of states prediction to direct Hamiltonian learning, ML-enabled methods now provide quantum-accurate electronic structure calculations for systems comprising hundreds of thousands of atoms with orders of magnitude speedup. The protocols and resources outlined in this article provide researchers with practical pathways to implement these advanced techniques, opening new frontiers for simulating complex materials at experimentally relevant scales. As benchmark datasets continue to expand and algorithms become more sophisticated, the integration of ML and DFT promises to unlock previously intractable problems in materials design and discovery.
The integration of Machine Learning (ML) with Density Functional Theory (DFT) represents a transformative advancement in computational materials research and drug development. This synergy addresses a critical bottleneck: the prohibitive computational cost of solving the Kohn-Sham equations, which has long constrained dynamical studies of complex phenomena at scale [3]. ML serves as a powerful force multiplier, augmenting the capabilities of researchers by providing orders-of-magnitude speedup while maintaining chemical accuracy, thereby freeing scientists to focus on higher-level analysis and strategic innovation [9] [10]. This paradigm shift is particularly impactful for applications requiring high-throughput screening, such as designing new catalysts, materials for energy storage, and pharmaceutical compounds, where traditional DFT approaches are computationally limited [3].
The core of this transformation lies in treating the Kohn-Sham equation as an input-output problem. Instead of performing explicit, costly DFT calculations, end-to-end ML models learn to map atomic structures directly to electronic properties and thermodynamic quantities [3]. This approach successfully bypasses the traditional computational hurdles, achieving linear scaling with system size with a small prefactor, making previously inaccessible studies of thousands of atoms over nanoseconds feasible [3]. For research professionals, this translates to accelerated discovery cycles and the ability to explore vast chemical spaces with unprecedented efficiency.
Core ML Methodologies and Quantitative Benchmarks
Architectural Frameworks for ML-DFT
Several sophisticated ML architectures have been developed to emulate and augment traditional DFT workflows, each with distinct advantages for specific research applications.
Deep Learning for Charge Density Prediction: A groundbreaking approach uses an end-to-end deep learning model that maps atomic structure directly to electronic charge density, which then serves as a descriptor for predicting other properties [3]. This method employs Atom-Centered AGNI Fingerprints to represent the structural and chemical environment of each atom in a translation, permutation, and rotation-invariant manner [3]. The model predicts the decomposition of atomic charge density in terms of Gaussian-type orbitals (GTOs), with the model learning the optimal basis set from data rather than using predefined basis functions [3].
Electronic Charge Density as Universal Descriptor: A universal framework utilizes electronic charge density as the sole input descriptor for predicting multiple material properties [11]. This approach leverages the Hohenberg-Kohn theorem, which establishes a one-to-one correspondence between ground-state wavefunctions and real-space electronic charge density [11]. The methodology converts 3D charge density data into image representations and employs Multi-Scale Attention-Based 3D Convolutional Neural Networks (MSA-3DCNN) to extract features and establish mappings to target properties [11].
Crystal Graph Convolutional Neural Networks (CGCNN): For crystalline materials, CGCNNs create a crystal graph representation where atoms serve as nodes and chemical bonds as edges, enabling accurate prediction of formation energies, band gaps, and elastic moduli [12].
Table 1: Performance Benchmarks of ML-DFT Models for Property Prediction
| Model Architecture | Target Properties | Accuracy Metrics | Computational Speedup | Applicable Systems |
|---|---|---|---|---|
| Deep Learning Charge Density Framework [3] | Charge density, DOS, potential energy, atomic forces, stress tensor | Chemical accuracy maintained | Orders of magnitude (linear scaling) | Organic molecules, polymer chains, crystals (C, H, N, O) |
| Universal Density-based MSA-3DCNN [11] | 8 different ground-state properties | Avg. R²: 0.66 (single-task), 0.78 (multi-task) | Not specified | Diverse crystalline materials |
| Graph Neural Networks (GNNs) [12] | Formation energy, band gaps, elastic moduli | Better than DFT accuracy reported | Hundreds of times faster than DFT | Crystalline materials |
Addressing Dataset Redundancy with MD-HIT
A critical challenge in ML for materials science is dataset redundancy, where highly similar materials in training sets lead to overestimated model performance and poor generalization [12]. The MD-HIT algorithm addresses this by controlling redundancy in material datasets, similar to CD-HIT in bioinformatics [12]. When applied to composition- and structure-based prediction problems, models trained on MD-HIT-processed datasets show relatively lower performance on standard test sets but better reflect true prediction capability for novel materials [12]. This is particularly important for real-world applications where discovering new functional materials requires extrapolation rather than interpolation [12].
Table 2: ML Model Performance with and without Redundancy Control
| Evaluation Scenario | Reported Formation Energy MAE (eV/atom) | Generalization Capability | Recommended Use Case |
|---|---|---|---|
| Random splitting (high redundancy) [12] | 0.064-0.07 | Overestimated, poor OOD performance | Preliminary screening |
| With redundancy control (MD-HIT) [12] | Relatively higher MAE | Better reflects true capability | Discovery of novel materials |
| K-fold Forward Cross-Validation [12] | Significantly higher MAE | Reveals weak extrapolation | Critical applications requiring robustness |
Experimental Protocols and Application Notes
Protocol: ML-DFT for Organic Materials
This protocol outlines the procedure for implementing a deep learning framework to emulate DFT calculations for organic materials containing C, H, N, and O, based on established methodologies [3].
Research Reagent Solutions
- Software Requirements: Vienna Ab Initio Simulation Package (VASP) for reference DFT calculations [3]
- Fingerprinting Tool: Atom-centered AGNI fingerprints for structure representation [3]
- ML Framework: Deep neural network implementation (Python with TensorFlow/PyTorch)
- Data Availability: Over 118,000 structures for training and testing [3]
Step-by-Step Procedure
Reference Data Generation:
- Perform DFT-based molecular dynamics (MD) runs at high temperatures (300K for molecules and polymer chains, 100-2500K for polymer crystals) to procure random snapshots with configurational diversity [3].
- Compute reference electronic properties (charge density, density of states, band properties) and atomic quantities (potential energy, atomic forces, stress tensor) using DFT [3].
Data Segmentation:
Atomic Fingerprinting:
Model Architecture Implementation:
- Implement a two-step learning procedure:
Reference System Transformation:
- Define an internal reference system using the two nearest neighbors of each atom [3].
- Calculate transformation matrices to convert from internal orthonormal reference systems to the global Cartesian system [3].
- Apply these transformations to rotation-variant properties like electron density and atomic forces [3].
Model Training and Validation:
- Train models using the preprocessed datasets.
- Validate prediction accuracy against DFT-calculated properties.
- Evaluate performance on independent test sets not used during training.
Protocol: Universal Property Prediction from Electronic Density
This protocol describes a methodology for predicting multiple material properties using electronic charge density as a universal descriptor [11].
Research Reagent Solutions
- Data Source: Materials Project CHGCAR files containing 3D charge density matrices [11]
- DL Architecture: Multi-Scale Attention-Based 3DCNN (MSA-3DCNN) [11]
- Preprocessing Tools: Data standardization and image representation pipelines [11]
Step-by-Step Procedure
Data Curation:
- Acquire CHGCAR files from Materials Project containing 3D charge density matrices [11].
- Collect corresponding target properties for training.
Data Standardization:
Model Configuration:
Training Approach:
Performance Validation:
Workflow Visualization
ML-DFT Two-Step Prediction Workflow
Implementation Considerations for Research Applications
Data Quality and Model Generalization
The performance of ML-DFT models heavily depends on data quality and diversity. Researchers must address several critical considerations:
Redundancy Control: Implement MD-HIT or similar algorithms to eliminate highly similar structures from training sets, ensuring models generalize to novel materials rather than merely interpolating between similar examples [12]. This is particularly crucial for applications in drug development where novel molecular entities are the target.
Multi-Task Learning: Leverage multi-task learning frameworks where possible, as they demonstrate improved accuracy (average R² increasing from 0.66 to 0.78 in universal density models) compared to single-task approaches [11]. This enhancement stems from the physical correlations between different material properties that the model can exploit.
Transferability Challenges: Recognize that excellent performance on benchmark datasets with random splitting does not guarantee success for out-of-distribution samples [12]. Employ leave-one-cluster-out cross-validation or forward cross-validation for more realistic performance assessment [12].
Force Multiplication in Research Teams
The integration of ML-DFT approaches serves as a true force multiplier for research teams in several dimensions:
Accelerated Discovery Cycles: By reducing computation time from hours/days to seconds/minutes for property prediction, ML-DFT enables rapid screening of candidate materials or molecular compounds [3] [10]. This allows research teams to explore significantly larger chemical spaces with the same resources.
Augmented Expertise: ML tools handle repetitive prediction tasks, freeing researchers to focus on higher-value activities such as experimental design, result interpretation, and hypothesis generation [9] [10]. This effectively extends the capabilities of each team member without requiring expansion of team size.
Democratization of Computational Tools: The speed and accessibility of ML-based property prediction make advanced computational screening available to smaller research groups that may lack resources for extensive DFT calculations [9].
Machine learning has unequivocally established itself as a force multiplier in computational materials research and drug development, transforming the traditional DFT workflow from a computational bottleneck to an efficient discovery engine. The frameworks described herein—from charge density-based prediction to universal property models—demonstrate that ML can achieve chemical accuracy with orders-of-magnitude speedup [3] [11]. However, critical challenges remain in ensuring model generalizability beyond benchmark datasets, particularly for novel material classes with limited training data [12].
Future advancements will likely focus on improving extrapolation capabilities, developing better uncertainty quantification methods, and creating more sophisticated multi-task learning architectures that capture the fundamental physics underlying material properties [12] [11]. As these technologies mature, the integration of ML-DFT will become increasingly central to materials validation research, enabling accelerated discovery and development across pharmaceuticals, energy storage, catalysis, and beyond. For research professionals, embracing these tools represents not merely an adoption of new technology, but a strategic transformation of the research paradigm itself.
The integration of Density Functional Theory (DFT) and Machine Learning (ML) is transforming the landscape of materials research by creating a powerful, iterative discovery loop. While DFT provides foundational quantum-mechanical calculations of material properties, it often faces challenges related to computational cost and accuracy for complex systems [5] [13]. Machine learning addresses these limitations by learning from existing DFT and experimental data to build predictive models, which in turn guide new DFT calculations and experimental validation [14] [15]. This synergistic integration significantly accelerates the discovery and optimization of electronic, mechanical, and catalytic materials, enabling researchers to navigate vast chemical spaces with unprecedented efficiency.
Application Notes and Protocols
The integration of DFT and ML has yielded significant advancements across various sub-disciplines of materials science. The table below summarizes key protocols, their technological impacts, and specific material systems targeted by these approaches.
Table 1: Key Application Areas of Integrated DFT and ML for Materials Discovery
| Application Area | DFT Contribution | ML Methodology | Key Outcome/Impact | Example Material Systems |
|---|---|---|---|---|
| Electronic Materials Band gap prediction [13] | High-fidelity band gap & lattice parameter calculations (DFT+U) as training data. | Supervised regression models (e.g., MLP) using Ud/f and Up as features. | Accurate, low-cost prediction of electronic properties; guides high-throughput screening. | Metal oxides (TiO₂, ZnO, CeO₂, ZrO₂) [13] |
| Mechanical/Structural Materials Phase stability [5] | Calculation of formation enthalpies (Hf) for alloys. | Neural network (MLP) to predict error between DFT and experimental enthalpies. | Improved reliability of phase diagram predictions for alloy design. | High-temp alloys (Al-Ni-Pd, Al-Ni-Ti) [5] |
| Catalytic Materials Acid-stability screening [16] | Evaluation of Pourbaix decomposition free energy (ΔGpbxOER) using HSE06. | SISSO-based symbolic regression to identify analytical descriptors. | Efficient identification of acid-stable oxides for electrocatalysis. | Water-splitting oxides [16] |
| High-Throughput Discovery General crystal stability [17] | Energetics of ~48,000 known crystals for model training and verification. | Scalable graph neural networks (GNoME) with active learning. | Order-of-magnitude expansion of known stable crystals. | Diverse inorganic crystals [17] |
Protocol for Electronic Materials: Band Gap Prediction in Metal Oxides
1. Objective: To accurately and efficiently predict the band gaps and lattice parameters of strongly correlated metal oxides by integrating DFT+U calculations with supervised machine learning [13].
2. Background: Standard DFT functionals (e.g., PBE) systematically underestimate the band gaps of metal oxides. The DFT+U method, which adds Hubbard corrections for on-site electron-electron interactions, improves accuracy but requires computationally expensive benchmarking to find optimal U parameters. A hybrid DFT+U+ML workflow overcomes this bottleneck [13].
3. Experimental Workflow:
- Step 1: High-Parameter DFT+U Calculations. Perform high-throughput DFT+U calculations for target metal oxides (e.g., rutile/anatase TiO₂, ZnO, CeO₂). The Hubbard U parameters are applied to both the metal d/f orbitals (U({d/f})) and the oxygen p orbitals (U(p)). A wide grid of integer (U(p), U({d/f})) pairs is explored to generate a comprehensive dataset of band gaps and lattice parameters [13].
- Step 2: Dataset Curation. The input features for the ML model are the U(p) and U({d/f}) values. The target outputs are the resulting band gaps and lattice parameters from the DFT+U calculations [13].
- Step 3: Model Training and Validation. Train relatively simple supervised ML regression models (e.g., Multi-layer Perceptrons/MLPs) on the generated dataset. The model learns to map the input (U(p), U({d/f})) values to the output structural and electronic properties. Model performance is evaluated using standard cross-validation techniques [13].
- Step 4: Prediction and Workflow Integration. The trained ML model can rapidly predict band gaps and lattice parameters for any given (U(p), U({d/f})) pair at a fraction of the computational cost of a full DFT+U calculation. This allows for rapid identification of optimal U parameters and pre-screening of materials before final, validation-level DFT calculations [13].
Protocol for Mechanical & Structural Materials: Alloy Phase Stability
1. Objective: To correct systematic errors in DFT-calculated formation enthalpies of alloys, thereby enabling reliable prediction of phase stability in binary and ternary systems [5].
2. Background: The predictive accuracy of DFT for alloy formation enthalpies is limited by intrinsic errors of exchange-correlation functionals. These errors are particularly detrimental for calculating ternary phase diagrams, where small energy differences determine stable phases. An ML-based correction model significantly improves agreement with experimental data [5].
3. Experimental Workflow:
- Step 1: Data Curation. Assemble a dataset containing both DFT-calculated and experimentally measured formation enthalpies (H(_f)) for a set of binary and ternary alloys. The dataset must be rigorously filtered to include only reliable experimental values [5].
- Step 2: Feature Engineering. Represent each material with a structured feature vector designed to capture chemical trends. This includes [5]:
- Elemental concentrations ((xA, xB, ...)).
- Weighted atomic numbers ((xA ZA, xB ZB, ...)).
- Interaction terms (e.g., (xA xB, xA xB x_C, ...)) to capture multi-element effects.
- Step 3: Error Learning and Model Training. Define the target variable as the discrepancy, ( \Delta Hf = Hf^{\text{exp}} - Hf^{\text{DFT}} ). Train a neural network model (e.g., a Multi-Layer Perceptron with three hidden layers) to learn the mapping from the feature vector to ( \Delta Hf ) [5].
- Step 4: Model Validation and Application. Validate the model using leave-one-out cross-validation (LOOCV) and k-fold cross-validation to ensure robustness and prevent overfitting. For a new material, the predicted ( \Delta Hf ) is added to its DFT-calculated ( Hf ) to obtain a corrected, more accurate formation enthalpy for phase stability assessment [5].
Protocol for Catalytic Materials: Identifying Acid-Stable Electrocatalysts
1. Objective: To efficiently identify oxide materials that are thermodynamically stable under acidic oxygen evolution reaction (OER) conditions using an active learning workflow guided by symbolic regression [16].
2. Background: Discovering earth-abundant, acid-stable oxides for water splitting is crucial for sustainable hydrogen production. Directly evaluating stability via Pourbaix decomposition free energy ((\Delta G_{pbx}^{OER})) using high-quality DFT-HSE06 calculations is computationally prohibitive for large material spaces. The SISSO (Sure-Independence Screening and Sparsifying Operator) method identifies analytical descriptors from a large feature space, making it ideal for capturing complex materials properties [16].
3. Experimental Workflow:
- Step 1: Initial Sampling and Feature Compilation. Start with an initial training set of ~250 oxides with HSE06-calculated (\Delta G_{pbx}^{OER}) values. For all materials in the search space, compile a wide range of primary features (e.g., elemental properties, composition-averaged orbital radii, standard deviation of oxidation states) [16].
- Step 2: Ensemble SISSO Model Training. Train an ensemble of SISSO models to obtain both predictions and uncertainty estimates. This involves [16]:
- Bagging with Monte-Carlo Dropout: Create multiple bootstrapped training datasets. For each set, randomly drop a subset (e.g., 20%) of the primary features before training a SISSO model.
- The ensemble's mean prediction and standard deviation provide the property value and associated uncertainty.
- Step 3: Active Learning for Data Acquisition. Use the ensemble model to screen a large pool of candidate oxides. Select materials for the next HSE06 calculation based on a criterion that balances exploitation (predicted (\Delta G_{pbx}^{OER}) is low/stable) and exploration (high prediction uncertainty). This efficiently targets the most promising and informative regions of the materials space [16].
- Step 4: Iteration and Discovery. Iterate the process by adding the new HSE06 data to the training set and retraining the ensemble SISSO model. This workflow identified 12 acid-stable oxides from a pool of 1470 in only 30 active learning iterations [16].
Successful implementation of integrated DFT+ML workflows relies on a suite of computational tools and data resources.
Table 2: Essential Research Toolkit for DFT and ML Materials Discovery
| Tool/Resource Category | Name/Example | Function/Purpose | Key Application in Workflow |
|---|---|---|---|
| Computational Databases | Materials Project (MP) [17] [15], OQMD [17] [15], AFLOW [17], ICSD [17] [15] | Sources of crystal structures and pre-computed properties for training and benchmarking. | Provides initial data for model training; source of candidate structures. |
| DFT Calculation Software | VASP [17] [13], FHI-aims [16] | Performs first-principles quantum mechanical calculations to determine energy, structure, and properties. | Generates high-quality training data and validates final candidate materials. |
| Machine Learning Frameworks | Graph Neural Networks [17], SISSO [16], Multi-layer Perceptrons [5] [13] | Learns complex patterns and structure-property relationships from data. | Core engine for predictive modeling and guiding active learning. |
| Validation & Benchmarking Tools | MatFold [18], k-fold & Leave-One-Out CV [5] | Provides standardized protocols to assess model generalizability and prevent over-optimistic performance estimates. | Critical for evaluating model robustness, especially for materials discovery tasks. |
Workflow Visualization
The following diagrams, generated using Graphviz DOT language, illustrate the logical flow of the key integrated DFT and ML protocols described in this document.
DFT+ML Workflow for Electronic Materials
ML Correction Protocol for Alloy Phase Stability
Active Learning Workflow for Stable Catalysts
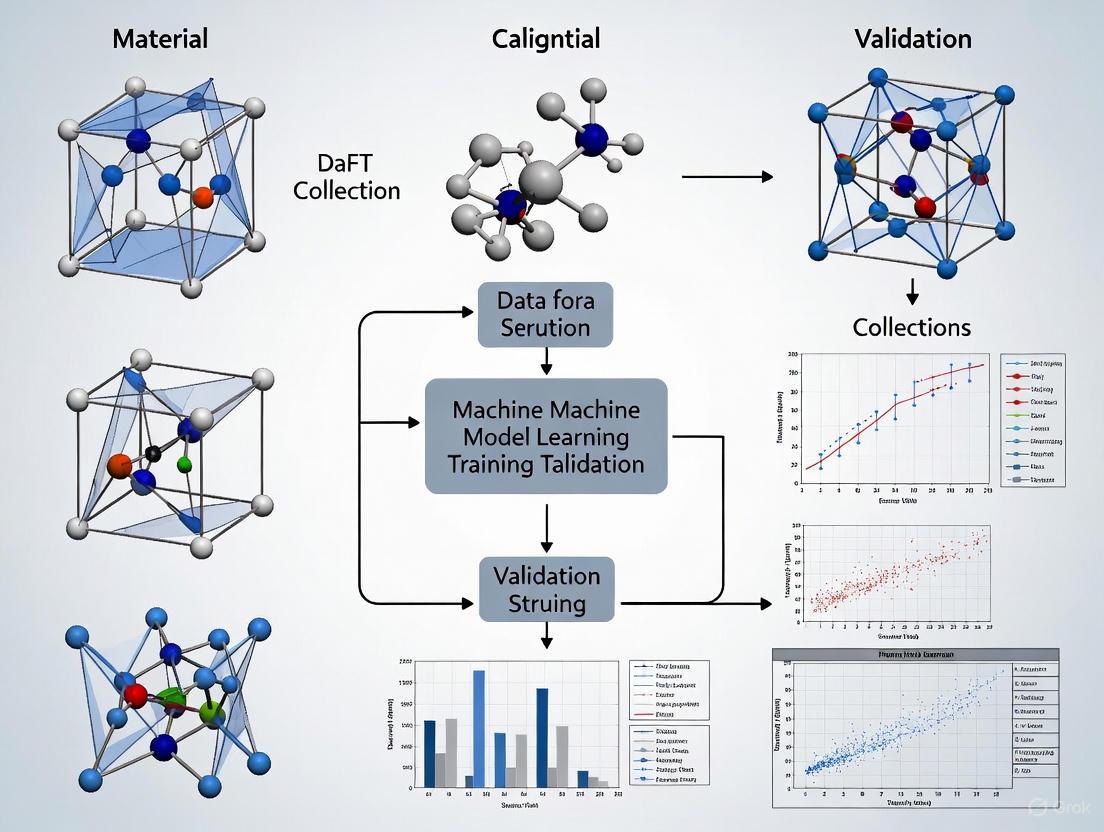
The integration of Density Functional Theory (DFT) and Machine Learning (ML) is revolutionizing materials science, enabling the rapid discovery and design of novel functional materials. This paradigm establishes a closed-loop workflow where computationally generated data trains ML models that predict material properties, design new candidates, and ultimately require experimental validation to confirm real-world utility. This framework is particularly vital in fields like battery research, catalyst design, and nanomaterial development, where traditional methods are often time-consuming and resource-intensive [14] [19] [20]. This document outlines the detailed protocols and application notes for implementing this workflow, providing researchers with a structured approach to accelerate materials validation research.
The Integrated DFT-ML Workflow: A Step-by-Step Breakdown
The general process for integrating DFT and machine learning in materials science involves several key stages, from initial data generation to the final experimental validation of predictions. The following diagram illustrates this comprehensive workflow and the logical relationships between its components:
Stage 1: DFT-Generated Training Data
Objective: To generate a reliable and comprehensive dataset of material properties for subsequent machine learning tasks.
Protocol 1.1: Performing High-Throughput DFT Calculations
- System Selection: Define the chemical space of interest (e.g., ternary alloys, perovskite oxides). For example, in studying alloys for high-temperature applications, focus on systems like Al-Ni-Pd and Al-Ni-Ti [5].
- Computational Setup: Utilize standardized settings to ensure consistency.
- Software: Employ plane-wave codes (VASP, Quantum ESPRESSO) or the Exact Muffin-Tin Orbital (EMTO) method [5].
- Exchange-Correlation Functional: Select an appropriate functional (e.g., Perdew-Burke-Ernzerhof (PBE) GGA) [5].
- k-point Mesh: Use a dense mesh (e.g., (17 \times 17 \times 17) for cubic systems) for Brillouin zone integration [5].
- Convergence Parameters: Strictly enforce convergence criteria for energy, force, and stress to ensure result reliability.
- Property Calculation: Calculate target properties such as:
- Formation enthalpy ((H_f)) using Equation 1 [5].
- Electronic band structure and density of states.
- Elastic constants and bulk modulus.
- Data Curation: Store calculated properties in structured databases (e.g., Materials Project, OQMD, AFLOW, NOMAD) with consistent metadata [19].
Stage 2: Data Preprocessing and Feature Engineering
Objective: To transform raw DFT data into a clean, well-characterized dataset suitable for machine learning.
Protocol 2.1: Data Preprocessing and Redundancy Control
- Handling Missing Data: Identify and address missing values, either by imputation or removal, depending on the dataset size and context.
- Data Transformation and Standardization: Apply normalization or standardization to feature sets to ensure uniform scaling, which improves the stability and convergence of many ML models [21].
- Redundancy Control with MD-HIT: To avoid over-optimistic performance estimates and ensure model generalizability, apply redundancy control algorithms.
- Rationale: Materials databases contain many highly similar structures due to historical "tinkering" in material design. Random splitting of data can lead to data leakage [12].
- Method: Use the MD-HIT algorithm to cluster materials based on composition or structural similarity. Ensure that no two highly similar materials are in both the training and test sets [12].
- Outcome: This provides a more realistic evaluation of a model's extrapolation capability and its potential to discover truly novel materials [12].
Protocol 2.2: Feature Engineering and Selection
- Descriptor Generation:
- Compositional Descriptors: Use tools like Mendeleev, Matminer, or Magpie to generate features from elemental properties (e.g., atomic number, electronegativity, ionic radii) and their statistical summaries (mean, range, standard deviation) [21].
- Structural Descriptors: For inorganic crystals, use universal descriptors [21]. For organic molecules, employ molecular fingerprints or descriptors from RDKit or PaDEL [21].
- Domain Knowledge Descriptors: Incorporate known physical descriptors, such as the tolerance factor for perovskites [21] or features for high-entropy alloy phase parameters [21].
- Text-Based Descriptors: As an innovative approach, automatically generate human-readable text descriptions of crystal structures (e.g., using Robocrystallographer) and use language models to process them [22].
- Feature Selection:
- Objective: Reduce overfitting and model complexity by identifying the most relevant feature subset.
- Methods: Employ a combination of:
Stage 3: Machine Learning Model Development
Objective: To train, evaluate, and select ML models that accurately map material features to target properties.
Protocol 3.1: Model Training and Evaluation with Robust Validation
- Model Selection: Choose from a diverse set of algorithms, which may include:
- Random Forests: For robust, interpretable models with good performance on small datasets [22].
- Graph Neural Networks (GNNs): For structure-based property prediction, as they naturally handle crystal graphs [19] [22].
- Transformer Models: For text-based representations of materials, which can offer high accuracy and interpretability [22].
- Neural Networks: Multi-layer perceptrons (MLPs) for learning complex, non-linear relationships in high-dimensional data [5].
- Robust Data Splitting: Avoid simple random splitting. Instead, use:
- Model Interpretation:
Table 1: Summary of Key ML Models and Their Applications in Materials Science
| Model Type | Best Suited For | Key Advantages | Considerations |
|---|---|---|---|
| Random Forest [22] | Small datasets, compositional trends | High interpretability, less prone to overfitting | Limited performance for complex structural dependencies |
| Graph Neural Networks [19] [22] | Structure-property relationships | State-of-the-art accuracy, natural for crystals | "Black-box" nature, requires large data, computationally intensive |
| Transformers (Text-Based) [22] | Small-data regimes, interpretability needs | High accuracy with text descriptions, explanations align with human rationale | Dependent on quality of text description |
| Multi-layer Perceptron [5] | Correcting DFT errors, non-linear mappings | Can learn complex patterns from diverse features | Can be prone to overfitting without careful regularization |
Stage 4: Prediction, Design, and Experimental Validation
Objective: To use trained ML models for the prediction of new materials and to validate these predictions experimentally.
Protocol 4.1: ML-Driven Material Design and Prediction
- Virtual Screening: Generate a large library of hypothetical material compositions and/or structures. Use the trained ML model to rapidly screen this library for candidates with desired properties [19].
- Inverse Design: Employ generative models like Generative Adversarial Networks (GANs) or Variational Autoencoders (VAEs) to directly generate novel material structures that satisfy target property constraints [19].
Protocol 4.2: Experimental Validation of Predictions
- Synthesis: Synthesize the top candidate materials predicted by the ML model using appropriate techniques (e.g., solid-state reaction, sol-gel, chemical vapor deposition).
- Characterization: Characterize the synthesized materials to confirm their structure, composition, and target properties using techniques such as:
- X-ray diffraction (XRD) for crystal structure.
- Electron microscopy (SEM/TEM) for morphology.
- X-ray photoelectron spectroscopy (XPS) for elemental composition.
- Performance Testing: Test the material in its intended application (e.g., as a battery cathode, catalyst, or photovoltaic cell) to verify that its performance matches predictions [23] [20].
- Collaboration: For computational groups, collaboration with experimentalists is often essential for this step [23].
- Use of Reference Materials: For nanomaterial characterization, use nanoscale Certified Reference Materials (CRMs) or Reference Test Materials (RTMs) to validate measurement instruments and protocols, ensuring reliability and supporting regulatory approval [24].
Table 2: Key Research Reagent Solutions for DFT-ML Workflows
| Item Name | Function/Application | Examples/Specifications |
|---|---|---|
| DFT Software | First-principles calculation of material properties. | VASP, Quantum ESPRESSO, EMTO-CPA [5] |
| Materials Databases | Source of training data (computational and experimental). | Materials Project [12], OQMD [12], JARVIS-DFT [22], PubChem [23] |
| Feature Extraction Tools | Generation of numerical descriptors from material composition/structure. | Matminer [21], RDKit [21], Mendeleev [21] |
| Text Description Generator | Creates human-readable crystal structure descriptions for ML. | Robocrystallographer [22] |
| Redundancy Control Tool | Clusters materials by similarity to prevent data leakage in model evaluation. | MD-HIT algorithm [12] |
| ML/AI Frameworks | Platform for building and training machine learning models. | TensorFlow, PyTorch, AutoGluon/TPOT for AutoML [19] |
| Nanoscale Reference Materials | Validation of characterization methods for nanomaterials. | NIST Gold Nanoparticles (e.g., NIST RM 8011, 8012, 8013) [24] |
Case Study: Correcting DFT Formation Enthalpies with Machine Learning
Background: A significant challenge in using DFT for predicting phase diagrams is the intrinsic error in formation enthalpy ((H_f)) calculations, which limits predictive accuracy for ternary systems [5].
Protocol: ML-based DFT Error Correction
- Data Collection:
- Compile a dataset of DFT-calculated formation enthalpies ((Hf^{DFT})) for a set of binary and ternary alloys (e.g., Al-Ni-Pd system).
- Assemble a corresponding set of experimentally measured formation enthalpies ((Hf^{Exp})) from reliable literature or databases. This serves as the ground truth [5].
- Target Variable Definition: The machine learning model is not trained to predict the formation enthalpy directly. Instead, it is trained to predict the error or discrepancy: (\Delta Hf = Hf^{Exp} - H_f^{DFT}) [5].
- Feature Engineering:
- Model Training and Implementation:
- Train a neural network (e.g., a Multi-layer Perceptron) as a regressor to learn the mapping:
Input Features ->(\Delta Hf) [5]. - Apply rigorous cross-validation (e.g., Leave-One-Out CV) to prevent overfitting [5].
- The corrected, more accurate formation enthalpy is then obtained as: (Hf^{Corrected} = Hf^{DFT} + \Delta Hf^{ML}).
- Train a neural network (e.g., a Multi-layer Perceptron) as a regressor to learn the mapping:
- Outcome: This approach significantly improves the accuracy of phase stability predictions, making DFT-based calculations more reliable for guiding the discovery of new alloys [5].
The workflow from DFT-generated data to ML-driven prediction and experimental validation represents a powerful, iterative engine for modern materials discovery. Success hinges on the meticulous execution of each stage: generating high-quality data, rigorously controlling for dataset redundancy, selecting appropriate models and features, and—most critically—closing the loop with experimental synthesis and validation. By adhering to the detailed protocols and utilizing the toolkit outlined in this document, researchers can robustly integrate computational and experimental efforts, thereby accelerating the development of next-generation functional materials.
Frameworks in Action: Implementing ML Force Fields and Predictive Models for Material Properties
In the study of two-dimensional twisted moiré materials, such as twisted bilayer graphene and transition metal dichalcogenides (TMDs), lattice relaxation profoundly influences electronic properties, including the emergence of strongly correlated states, unconventional superconductivity, and Mott insulating states [25]. However, accurately modeling these relaxation effects presents a significant computational challenge. Traditional density functional theory (DFT) calculations, while accurate, scale cubically with the number of atoms, making them prohibitively expensive for moiré superlattices containing thousands of atoms, especially at small twist angles where the supercell size becomes enormous [25]. While empirical force fields and parameterized continuum models offer alternatives, they often lack the accuracy or transferability required for predictive simulations [25].
Machine learning force fields (MLFFs) have emerged as a powerful solution to this computational bottleneck, capable of predicting energies and forces with near-DFT accuracy at a fraction of the computational cost. The DPmoire software package represents a specialized tool designed specifically for constructing accurate MLFFs in moiré systems [25] [26]. This application note details the implementation, validation, and application of DPmoire within a broader research framework integrating DFT and machine learning for material validation, providing researchers with comprehensive protocols for deploying this tool in the study of complex moiré materials.
Scientific Rationale: Why Specialized MLFFs for Moiré Systems?
Moiré structures exhibit unique characteristics that necessitate specialized approaches for force field development. In twisted bilayers, the varying local atomic registries create a complex potential energy landscape where different regions (AA, MX, XM in TMDs) correspond to distinct stacking configurations with different energy states [25]. The energy scales of electronic bands in these systems are often on the order of millielectronvolts (meV), comparable to the accuracy limits of universal MLFFs [25]. This precision requirement demands MLFFs specifically tailored to individual material systems rather than relying on general-purpose models.
The DPmoire methodology leverages the physical insight that at minimal twist angles, local atomic configurations in moiré structures closely resemble those in non-twisted systems with different stacking registries [25]. By comprehensively sampling the potential energy surfaces of these non-twisted configurations, DPmoire effectively reconstructs the potential energy landscape of twisted structures, enabling accurate and efficient relaxation of moiré superlattices.
Table: Comparison of Computational Methods for Moiré System Relaxation
| Method | Computational Scaling | Accuracy | Applicability to Small Twist Angles |
|---|---|---|---|
| Standard DFT | O(N³) | High | Limited |
| Continuum Models | O(1) | Moderate | Excellent |
| Empirical Force Fields | O(N) | Variable | Good |
| Universal MLFFs | O(N) | Moderate-High | Good |
| DPmoire (Specialized MLFF) | O(N) | High | Excellent |
DPmoire Architecture and Workflow
DPmoire is structured into four functional modules that streamline the process of generating, training, and validating MLFFs for moiré systems [26]:
DPmoire.preprocess: Automatically combines layer structures and generates shifted structures of a 2×2 supercell, prepares twisted structures for test sets, and manages VASP input files based on provided templates.DPmoire.dft: Submits VASP calculation jobs through the Slurm workload manager.DPmoire.data: Collects DFT-calculated data fromML_ABandOUTCARfiles, then generates training and test set files inextxyzformat compatible with Allegro and NequIP packages.DPmoire.train: Modifies system-dependent settings in configuration files and submits training jobs for Allegro or NequIP MLFFs.
The software utilizes advanced E(3)-equivariant graph neural network algorithms, specifically NequIP and Allegro, which ensure covariance among inputs, outputs, and hidden layers, leading to enhanced data efficiency and model accuracy [25]. For systems where robust empirical potentials are scarce, DPmoire provides a systematic approach to generating accurate MLFFs.
Research Reagent Solutions: Essential Tools for Moiré MLFF Development
Table: Essential Software Tools for DPmoire Implementation
| Tool Name | Function | Implementation Notes |
|---|---|---|
| DPmoire | Core package for generating and training MLFFs for moiré systems | Requires pre-installation of NequIP or Allegro for training [26] |
| VASP | Ab initio electronic structure calculations for dataset generation | Must be properly licensed and configured [26] |
| NequIP | E(3)-equivariant graph neural network for MLFF training | Provides high data efficiency [25] |
| Allegro | E(3)-equivariant MLFF algorithm optimized for large structures | Suitable for parallel computing [25] |
| Slurm | Workload manager for job submission and management | Essential for HPC environments [26] |
Application Notes: MX₂ Moiré Systems
DPmoire has been successfully applied to develop MLFFs for MX₂ materials (M = Mo, W; X = S, Se, Te), demonstrating robust performance in replicating electronic and structural properties obtained from DFT relaxations [25]. The MLFFs were rigorously validated against standard DFT results, confirming their efficacy in capturing complex atomic interactions within these layered materials [25].
For twisted TMD systems, the lattice relaxation significantly modulates the moiré potential, which in turn affects the electronic band structures. Experimental studies using scanning tunneling microscopy (STM) have documented relaxation patterns in TMDs resulting from lattice reconstruction [25], providing validation for computational approaches. DPmoire-generated MLFFs enable researchers to efficiently explore these relaxation effects across different twist angles without the computational burden of direct DFT calculations.
Table: Performance Metrics of DPmoire-Generated MLFFs for MX₂ Materials
| Material System | Energy RMSE (eV/atom) | Force RMSE (eV/Å) | Stress RMSE (kBar) | Twist Angle Range |
|---|---|---|---|---|
| MoS₂ | 1.21×10⁻⁴ | 8.13×10⁻³ | 6.08×10⁻¹ | 3.89° and smaller [25] |
| WS₂ | Data not specified in sources | Similar performance expected | Similar performance expected | 3.89° and smaller |
| MoSe₂ | Data not specified in sources | Similar performance expected | Similar performance expected | 3.89° and smaller |
| WSe₂ | Data not specified in sources | Similar performance expected | Similar performance expected | 3.89° and smaller |
Experimental Protocols
Dataset Generation Protocol
Initial Setup: Prepare
top_layer.poscarandbot_layer.poscarfiles ensuring the c-axis is sufficiently large. Create INCAR templates (init_INCAR,rlx_INCAR,MD_INCAR,MD_monolayer_INCAR,val_INCAR) with appropriate van der Waals correction settings [26].Configuration File Preparation: Set up
config.yamlwith key parameters including:n_sectors: Number of grid points for in-plane shifts (typically 9×9 structures before symmetry reduction)symm_reduce: True (to reduce computational cost by leveraging crystal symmetry)twist_val: True (to generate twisted structures for validation)min_val_nandmax_val_n: Define range of twist angles (n=1: 21.97°, n=2: 13.17°, n=3: 9.43°, etc.)d: Initial interlayer distance [26]
Structure Generation: Execute
DPmoire.preprocessto generate shifted and twisted structures. The module automatically creates a 2×2 supercell with various stacking configurations and prepares twisted structures for validation sets.DFT Calculations: Run
DPmoire.dftto submit VASP calculations through Slurm. EnableVASP_ML=Truein configuration to use VASP's on-the-fly MLFF for efficient data generation [26].
MLFF Training and Validation Protocol
Data Collection: Use
DPmoire.datato compile DFT data fromML_ABandOUTCARfiles intoextxyzformat for Allegro/NequIP training.Model Training: Execute
DPmoire.trainto train the MLFF using NequIP or Allegro. Critical training parameters include:validation_dataset_file_name: Location of validation datasetn_val: Number of validation structures- Training iterations until F_{rmse} reaches below 1×10⁻² eV/Å [26]
Error Analysis: Perform rigorous training-set and test-set error analysis following VASP MLFF protocols [27]. Compare:
- Training-set error: RMSE between DFT reference and MLFF prediction on training data
- Test-set error: RMSE on external test set not used during training
- Ideal scenario: Both errors are similarly low [27]
Hyperparameter Optimization: Adjust MLFF parameters based on error analysis:
- If overfitting occurs (low training error, high test error), increase training structures or tune hyperparameters
- For high training and test errors, expand dataset diversity or adjust model architecture [27]
Performance Optimization and Troubleshooting
Incongruent Data Issues: If training loss decreases slowly or F_{rmse} struggles to reach below 1×10⁻² eV/Å:
- Verify consistent INCAR template settings (e.g., avoid using different vdW corrections for relaxation and MD simulations)
- Check temperature stability during MD simulations as VASP MLFF can become unstable with strange structures [26]
Computational Efficiency:
Species Handling: For atoms in different environments (e.g., surface vs bulk), consider treating them as separate species in POSCAR to improve accuracy, despite increased computational cost [28].
DPmoire represents a specialized computational tool that effectively addresses the unique challenges of modeling moiré materials by combining the accuracy of DFT with the efficiency of machine learning force fields. Its structured workflow—from dataset generation through model training to validation—provides researchers with a robust framework for investigating relaxation effects in twisted two-dimensional materials. The integration of E(3)-equivariant graph neural networks ensures high data efficiency and accuracy, making it possible to explore complex moiré systems with minimal computational overhead. As research in quantum materials continues to emphasize the importance of moiré engineering, tools like DPmoire will play an increasingly vital role in accelerating the discovery and understanding of novel material properties in these systems.
The integration of Density Functional Theory (DFT) and machine learning (ML) has ushered in a new paradigm in computational materials science, extending predictive capabilities beyond traditional calculations of energies and forces. While DFT provides a quantum mechanical foundation for understanding materials at the atomic scale, its computational demands and systematic errors have limited its effectiveness for predicting critical functional properties like band gaps and elastic moduli. These properties are essential for designing materials for specific applications in electronics, energy storage, and drug development, where accurate prediction of electronic and mechanical behavior is crucial. The emergence of ML approaches has created opportunities to overcome these limitations, enabling high-accuracy prediction of complex properties while significantly reducing computational costs.
This integration represents a fundamental shift from purely physics-based modeling to hybrid approaches that leverage data-driven insights. Where DFT calculations provide the foundational data, ML models learn the complex relationships between material composition, structure, and properties, allowing for rapid screening and discovery of novel materials. This partnership has proven particularly valuable for properties that are computationally expensive to calculate directly or that suffer from systematic errors in DFT approximations. As research in this field accelerates, standardized protocols and application notes are needed to guide researchers in implementing these powerful methods effectively.
Quantitative Performance of DFT-ML Methods for Critical Property Prediction
Table 1: Performance Metrics of ML Models for Band Gap Prediction
| Material System | ML Method | Prediction Target | Performance Metrics | Reference |
|---|---|---|---|---|
| Spinel Oxides (AyB1-y[AxB2-x]O4) | Composition-based ML | Band Gap | Accurate predictions based solely on compositions | [29] |
| Organic Molecules | Random Forest | HOMO/LUMO Energies | MAE: 0.15 eV (HOMO), 0.16 eV (LUMO) | [30] |
| Diverse Crystals | Universal ML Framework (Electronic Density) | Multiple Properties | R² up to 0.94 for various properties | [11] |
| RbCdF3 under stress | DFT Analysis | Band Gap Changes | Increase from 3.128 eV to 3.533 eV under stress (12% rise) | [31] |
Table 2: Performance in Predicting Elastic Properties
| Material System | ML Method | Prediction Target | Performance Metrics | Reference |
|---|---|---|---|---|
| Inorganic Crystals | ElaTBot-DFT (LLM) | Elastic Constant Tensor | 33.1% error reduction vs. domain-specific LLM | [32] |
| 2D Elastic Metamaterials | XGBoost | Band Gap Position & Bandwidth | MAE: 339.06 (position), 116.45 (bandwidth) | [33] |
| Binary/Ternary Alloys | Neural Network (MLP) | Formation Enthalpy | Improved accuracy over standard DFT | [34] |
| Diverse Materials | Multi-Scale Attention-Based 3DCNN | 8 Different Properties | R²: 0.66 (single-task), 0.78 (multi-task) | [11] |
Methodological Protocols for Property Prediction
Protocol 1: Band Gap Prediction for Spinel Oxides Using Composition-Based ML
Objective: To predict electronic conductivity and band gaps of spinel oxides (AyB1−y[AxB2−x]O4) using machine learning based solely on material composition.
Materials and Computational Requirements:
- Dataset: 190 different ternary spinel oxides with varied stoichiometries
- DFT Methods: Geometry optimization and band structure calculations
- ML Algorithms: Models trained on compositional features
- Validation: Comparison with experimental trends for manganese cobalt spinels
Step-by-Step Procedure:
- Database Construction: Perform DFT calculations on diverse spinel oxide compositions to generate a comprehensive dataset including band structure and conductivity properties [29].
Feature Engineering: Develop compositional descriptors based solely on element types and stoichiometries without structural information.
Model Training: Train machine learning algorithms to predict electronic conductivity and band gaps using the DFT-calculated database as training data.
Band Structure Fitting: Fit DFT-calculated band structures to tight-binding Hamiltonians for efficient electronic transport calculations.
Current Calculation: Compute current under 1V bias for each composition using Non-Equilibrium Green's Function (NEGF) and Landauer formalism.
Model Validation: Validate predictions against experimental trends, particularly for systems with high nickel content and manganese cobalt spinels.
Prediction: Deploy trained models to predict band gaps and conductivity for new spinel compositions not included in the original dataset.
Troubleshooting Tips:
- Magnetic moment anomalies may require re-running DFT optimization with new initial magnetic moments
- For wide bands that are difficult to fit to tight-binding Hamiltonians, consider alternative fitting approaches
- Ensure dataset diversity to cover a representative portion of possible compositional combinations
Protocol 2: Elastic Constant Tensor Prediction Using Large Language Models
Objective: To predict full elastic constant tensors of materials using domain-specific Large Language Models (LLMs) fine-tuned on computational and experimental data.
Materials and Computational Requirements:
- Base LLM: Llama2-7b model
- Training Data: Elastic constant data from Materials Project and other sources
- Structural Descriptors: robocrystallographer for text-based structural descriptions
- Compositional Information: Pymatgen for extracting compositional features
Step-by-Step Procedure:
- Data Curation: Collect and preprocess elastic constant tensor data from available databases, noting the scarcity of complete tensor data compared to other material properties [32].
Textual Representation: Convert crystal structures into text descriptions using robocrystallographer to create natural language representations suitable for LLM processing.
Feature Integration: Combine structural text descriptions with compositional information from Pymatgen to create comprehensive input prompts.
Model Fine-Tuning: Fine-tune the base Llama2-7b model on the formatted material property data to create the specialized ElaTBot-DFT model.
Retrieval-Augmented Generation (RAG): Implement RAG capabilities to enhance predictions without retraining by leveraging external tools and databases.
Validation: Evaluate model performance using hold-out test sets and compare against traditional ML approaches and other domain-specific LLMs.
Prediction: Deploy the fine-tuned model for elastic constant tensor prediction, bulk modulus calculation, and generation of new materials with targeted elastic properties.
Key Advantages:
- Reduces prediction errors by 33.1% compared to domain-specific materials science LLMs
- Enables direct prediction of full elastic constant tensors, not just individual components
- Supports multi-task learning for simultaneous prediction of multiple properties
- Allows natural language interaction, lowering barriers for non-specialists
Protocol 3: Universal Material Property Prediction Using Electronic Charge Density
Objective: To predict eight different material properties using a unified machine learning framework based solely on electronic charge density as a universal descriptor.
Materials and Computational Requirements:
- Descriptor: Electronic charge density from DFT calculations
- Deep Learning Model: Multi-Scale Attention-Based 3D Convolutional Neural Network (MSA-3DCNN)
- Dataset: CHGCAR files from Materials Project database
- Computational Framework: Python with deep learning libraries (PyTorch/TensorFlow)
Step-by-Step Procedure:
- Data Acquisition: Curate electronic charge density data from the Materials Project database, representing results as three-dimensional matrices in CHGCAR files [11].
Data Standardization: Address dimensional variations in charge density data across different materials using interpolation schemes to create unified representations.
Image Representation: Convert 3D charge density matrices into 2D image snapshots along different crystal directions while preserving spatial relationships.
Model Architecture: Implement MSA-3DCNN with attention mechanisms to extract relevant features from charge density representations.
Training Strategy: Employ both single-task and multi-task learning approaches, comparing their relative performance for different properties.
Feature Extraction: Leverage the model's ability to capture subtle local variations in electron density, including accumulation near chemical bonds.
Validation: Evaluate transferability across different material classes and property types, assessing true extrapolation capability.
Key Insights:
- Electronic charge density serves as a physically grounded universal descriptor according to the Hohenberg-Kohn theorem
- Multi-task learning significantly enhances prediction accuracy compared to single-task approaches (R²: 0.78 vs 0.66)
- The method demonstrates excellent transferability across different property types
- Model captures complex structure-property relationships without explicit feature engineering
Workflow Visualization: Integrating DFT and ML for Property Prediction
DFT-ML Integration Workflow for Property Prediction
Table 3: Essential Computational Tools for DFT-ML Integration
| Tool/Resource | Type | Primary Function | Application Example |
|---|---|---|---|
| VASP | DFT Software | Electronic structure calculations | Charge density calculation for ML descriptors [11] |
| Pymatgen | Python Library | Materials analysis | Compositional feature extraction [32] |
| robocrystallographer | Text Generation Tool | Structural description | Converting crystal structures to text for LLMs [32] |
| Materials Project | Database | Curated material properties | Source of training data for ML models [12] [11] |
| ElaTBot-DFT | Specialized LLM | Elastic property prediction | Predicting full elastic constant tensors [32] |
| MSA-3DCNN | Deep Learning Model | Charge density analysis | Universal property prediction [11] |
| XGBoost | ML Algorithm | Regression & Classification | Band gap prediction in metamaterials [33] |
Challenges and Future Perspectives
Despite significant progress, several challenges remain in the integration of DFT and ML for predicting critical material properties. Dataset redundancy represents a fundamental issue that can lead to overestimated model performance. Recent studies have shown that materials datasets often contain many highly similar materials due to historical tinkering approaches in material design [12]. This redundancy causes random splitting in ML model evaluation to fail, leading to over-optimistic performance metrics that don't reflect true predictive capability for novel materials. The MD-HIT algorithm has been developed to address this issue by reducing dataset redundancy, providing more realistic assessment of model performance.
The transferability of ML models across different material classes and properties remains another significant challenge. While traditional ML approaches have focused on predicting specific properties, recent advances in universal frameworks using electronic charge density show promise for multi-property prediction [11]. The electronic charge density serves as a physically grounded descriptor that contains comprehensive information about material behavior, enabling prediction of multiple properties within a unified framework.
Future developments will likely focus on improving model interpretability, addressing data scarcity for certain property types, and enhancing generalization to truly novel materials not represented in training data. The integration of large language models presents an exciting direction for materials science, offering natural language interfaces that lower barriers for non-specialists while providing powerful predictive capabilities [32]. As these methods mature, standardized protocols and benchmarking datasets will be essential for comparing different approaches and driving the field forward.
The synergy between DFT and machine learning continues to redefine the landscape of materials property prediction, moving beyond traditional limitations of energy and force calculations to enable accurate prediction of functionally critical properties like band gaps and elastic moduli. By combining physical principles with data-driven insights, this integrated approach promises to accelerate materials discovery and design across diverse applications from electronics to drug development.
The integration of Density Functional Theory (DFT) and machine learning (ML) has ushered in a transformative era for computational materials science, enabling the rapid prediction of material properties and the discovery of novel compounds. Traditional DFT calculations, while invaluable, are often hampered by high computational costs and systematic errors, particularly in complex systems with strong electron correlations [5] [13]. Machine learning, especially models utilizing Graph Neural Networks (GNNs), addresses these limitations by learning directly from the atomic structure of materials, offering a powerful, data-driven approach to complement first-principles calculations [35] [36]. The inherent graph-like nature of crystalline materials, where atoms naturally represent nodes and bonds represent edges, makes GNNs an exceptionally suitable architecture for modeling materials [35] [37].
Recent advancements have pushed beyond simple atom-bond representations by incorporating higher-order interactions. The inclusion of four-body interactions, such as dihedral angles, represents a significant leap in capturing the complex, multi-scale physics that govern material behavior [38]. These interactions are crucial for accurately describing the potential energy surface and for predicting sophisticated properties that depend on the precise spatial arrangement of atoms. Models like CrysGNN and CrysCo are at the forefront of this innovation, leveraging these advanced architectural principles to achieve state-of-the-art accuracy in property prediction [38]. This application note details the protocols for implementing these advanced GNN architectures, positioning them within a broader research framework for the validation of materials, particularly for applications in drug development and nanotechnology.
Table 1: Core Descriptors for Advanced GNN Models in Materials Science
| Descriptor Type | Specific Examples | Physical/Chemical Information Captured | Role in Model Architecture |
|---|---|---|---|
| Atomic Features | Group number, period number, electronegativity, atomic radius [35] | Element-specific chemical identity and properties | Node feature initialization |
| Two-Body Interactions | Interatomic bond lengths, bond types [35] | Pairwise atomic interactions, bond strength | Basic edge construction in atom graph |
| Three-Body Interactions | Bond angles (θ) [35] | Local atomic geometry, orbital hybridization | Enhanced edge features, local curvature |
| Four-Body Interactions | Dihedral angles (φ), torsional potentials [38] | Out-of-plane torsion, complex conformational energies | Critical for capturing periodicity and long-range interactions |
Advanced GNN Architectures and Quantitative Performance
The evolution of GNNs for crystal materials has progressed from capturing basic connectivity to modeling intricate geometric relationships. Initial models like CGCNN (Crystal Graph Convolutional Neural Network) laid the groundwork by representing crystals as graphs with atoms as nodes and bonds as edges [38]. Subsequent models like ALIGNN (Atomistic Line Graph Neural Network) improved accuracy by explicitly incorporating bond angles (three-body interactions) by constructing an additional graph from the bonds of the original atom graph [35] [38]. The latest generation of models, including CrysCo, now integrates four-body interactions, allowing them to capture an even more complete picture of the atomic environment, which is vital for predicting properties sensitive to complex structural deformations [38].
Quantitative benchmarking on large-scale public databases such as the Materials Project and JARVIS-DFT demonstrates the superior performance of these advanced architectures. For instance, the CGGAT (Crystal Gated Graph Attention Network) model, which combines a gated mechanism with an attention mechanism to weight the importance of different atomic neighbors, has been shown to outperform other GNN algorithms across a range of prediction tasks [35]. The following table summarizes the performance of several leading models, illustrating the gains achieved by incorporating more complex geometric information.
Table 2: Benchmarking Performance of Advanced GNN Models on Materials Project Data
| Model Architecture | Key Interactions Captured | Formation Energy (MAE in meV/atom) | Band Gap (MAE in eV) | Bulk Modulus (MAE in GPa) |
|---|---|---|---|---|
| CGCNN [38] | Two-body | ~28 | ~0.39 | ~0.078 |
| ALIGNN [38] | Two-body, Three-body | ~22 | ~0.32 | ~0.066 |
| MEGNet [38] | Two-body, Global state | ~21 | ~0.31 | ~0.068 |
| CGGAT [35] | Two-body, Three-body, Attention | ~19 | ~0.29 | ~0.063 |
| CrysCo [38] | Two-, Three-, and Four-body | ~17 | ~0.27 | ~0.059 |
The integration of DFT and ML is not limited to property prediction. Frameworks like GNoME (Graph Networks for Materials Exploration) from Google DeepMind use GNNs to discover new materials on an unprecedented scale, actively learning from DFT calculations to predict material stability and propose novel, synthesizable crystals [37]. This demonstrates a powerful闭环 (closed-loop) workflow where ML massively accelerates the discovery process, which is then validated by high-fidelity DFT.
Experimental and Computational Protocols
Protocol 1: DFT Data Generation for Training and Validation
Objective: To generate a high-quality, consistent dataset of crystal structures and their corresponding properties using DFT, which will serve as the ground truth for training and validating the GNN models.
- System Selection: Define the scope of the study (e.g., binary/ternary alloys, metal-organic frameworks, specific metal oxides). For example, studies may focus on systems like Al-Ni-Pd or Al-Ni-Ti for high-temperature applications, or metal oxides like TiO2 and ZnO for electronic properties [5] [13].
- Structure Acquisition & Preparation: Obtain crystal structures from databases like the Materials Project [35], OQMD [36], or COD. Standardize structures by refining atomic coordinates and lattice parameters.
- DFT Calculation Setup:
- Software: Employ plane-wave codes such as VASP [13] or EMTO [5].
- Exchange-Correlation Functional: Select an appropriate functional (e.g., PBE-GGA [5] [13]). For strongly correlated systems (e.g., metal oxides), apply the DFT+U method with Hubbard parameters (Ud/f, Up) to correct for self-interaction error [13].
- Computational Parameters: Ensure convergence of key parameters:
- Plane-wave kinetic energy cutoff: ≥ 520 eV.
- k-point mesh: Use a Γ-centered grid with a density of at least 1000/Å⁻³ per atom [5].
- Energy and force convergence criteria: Set to 10⁻⁶ eV and 0.01 eV/Å, respectively.
- Property Calculation: Calculate target properties for each structure. Key properties include:
- Formation Energy: Using Equation (1) from the background, comparing the total energy of the compound to its constituent elements in their standard states [5].
- Electronic Band Gap.
- Elastic Constants (for bulk and shear moduli).
- Data Curation: Filter the results to exclude calculations with poor convergence or missing data. Compile a final dataset of
(crystal structure, target property)pairs.
Protocol 2: Implementing a GNN with Four-Body Interactions
Objective: To construct, train, and evaluate a GNN model capable of learning from the atomic structure and multi-body interactions to predict material properties.
- Graph Representation Construction:
- Atom Graph: Represent the crystal structure as a graph
G = (V, E), where nodesv_i ∈ Vrepresent atoms, and edgese_ij ∈ Erepresent bonds between atoms within a specified cutoff radius (e.g., 5-8 Å). Node features include atomic number, group, period, etc. Edge features include bond length and bond type [35]. - Edge Graph (for higher-order interactions): To model three- and four-body interactions, create a line graph
L(G)where nodes correspond to the edges inG(i.e., bonds), and edges inL(G)connect two bonds that share an atom (for angles) or form a dihedral (for four-body terms). The features of nodes inL(G)can be the original bond features, while edges inL(G)can be parameterized by the bond angle or dihedral angle [35] [38].
- Atom Graph: Represent the crystal structure as a graph
- Model Architecture (e.g., CrysCo, CGGAT):
- Input Layers: Embed atomic numbers and other scalar features into a continuous vector space.
- Message Passing Layers: Implement a series of graph convolution layers that operate on both the atom graph
Gand the edge graphL(G). For example, use Gated Graph Convolution (GatedGCN) or Graph Attention (GAT) layers [35]. The message passing should alternate between updating atom features based on their neighbors and updating bond/angle features. - Four-Body Integration: Specifically incorporate dihedral angle information, either as an additional edge feature in the edge graph or through a dedicated geometric learning module [38].
- Readout/Pooling Layer: Aggregate the final updated node/edge features into a fixed-dimensional global graph representation. This can be done via a mean/sum pool or an attention-based weighted sum [35].
- Output Layer: Pass the global representation through fully connected layers to predict the target property.
- Training Loop:
- Loss Function: Use Mean Absolute Error (MAE) or Mean Squared Error (MSE) between predictions and DFT-calculated labels.
- Optimization: Use the Adam optimizer with an initial learning rate of 10⁻³ to 10⁻⁴ and a batch size of 32 to 128.
- Validation & Regularization: Employ k-fold cross-validation or a hold-out test set. Use techniques like learning rate scheduling and early stopping to prevent overfitting [5].
- Model Evaluation: Evaluate the trained model on the test set and report standard metrics (MAE, RMSE, R²). Perform ablation studies to quantify the contribution of four-body interactions to the final predictive performance.
Successful implementation of the protocols above relies on a suite of software tools, datasets, and computational resources. The following table acts as a checklist for researchers embarking on projects integrating DFT and GNNs.
Table 3: Essential Research Reagents and Resources for DFT-GNN Integration
| Category | Item | Specific Examples & Citations | Primary Function |
|---|---|---|---|
| Computational Software | DFT Calculation Suites | VASP [13], EMTO [5], Quantum ESPRESSO | Perform first-principles electronic structure calculations. |
| Machine Learning Frameworks | PyTorch, TensorFlow, PyTorch Geometric (PyG), Deep Graph Library (DGL) | Build, train, and deploy GNN models. | |
| Data Resources | Crystal Structure Databases | Materials Project [35] [13], JARVIS-DFT [35], OQMD [36], COD | Provide initial crystal structures and pre-computed properties for training. |
| Pre-trained Models & Code | CrysGNN [38], ALIGNN [38], CGGAT [35], GNoME [37] (via GitHub repos) | Offer foundational models for transfer learning and benchmarking. | |
| Methodological Components | Exchange-Correlation Functionals | PBE, rPBE, PBEsol, HSE [13] | Define the approximation for electron interactions in DFT. |
| Hubbard U Corrections | DFT+U (Ud, Up) [13] | Improve DFT accuracy for strongly correlated electrons. | |
| Geometric Descriptors | Bond lengths (2-body), angles (3-body), dihedrals (4-body) [35] [38] | Encode local and medium-range atomic environment for GNNs. |
The integration of density functional theory (DFT) and machine learning (ML) has emerged as a powerful paradigm for accelerating materials discovery. However, the conventional pre-training and fine-tuning approach often struggles with the inherent diversity and disparity of material property prediction tasks. The MoMa (Modular framework for Materials) framework addresses these challenges by introducing a modular deep learning strategy that first trains specialized modules across a wide range of tasks and then adaptively composes synergistic modules tailored to each downstream scenario [39]. Evaluation across 17 datasets demonstrates MoMa's superiority, with a substantial 14% average improvement over the strongest baseline [39]. This protocol details the application of MoMa within a research context focused on integrating DFT and machine learning for material validation.
MoMa is designed to overcome two fundamental challenges in material property prediction: the diversity of material systems (e.g., crystals, molecules) and properties (e.g., electronic, mechanical), and the disparity in the physical laws governing these properties, which can lead to knowledge conflicts in unified models [39]. The framework operates through two major stages: Module Training & Centralization, and Adaptive Module Composition.
Core Components and Workflow
The following diagram illustrates the end-to-end workflow of the MoMa framework, from module training to deployment for a downstream prediction task.
Experimental Protocols
Module Training and Centralization Protocol
Objective
To train specialized, transferrable modules on high-resource material property prediction tasks and centralize them within the MoMa Hub repository [39].
Materials and Software Requirements
Table 1: Essential Research Reagents and Computational Tools
| Item | Function/Description | Implementation Notes |
|---|---|---|
| Pre-trained Backbone Encoder (e.g., CGCNN, MEGNet) | Base model for feature extraction from material structures [39]. | Provides initialization for all modules. Model-agnostic. |
| High-Resource Material Datasets | Training data for module specialization (e.g., from Matminer) [39]. | Requires >10,000 data points. Span thermal, electronic, mechanical properties. |
| Adapter Layers (Parameter-Efficient) | Lightweight neural networks inserted into backbone; alternative to full fine-tuning [39]. | Reduces GPU memory cost. Frozen backbone, only adapters updated. |
| MoMa Hub Repository | Centralized storage for trained module parameters [39]. | Enables knowledge reuse and privacy-aware contributions. |
Step-by-Step Procedure
- Backbone Initialization: Select a pre-trained material model (e.g., a graph neural network trained on crystal structures) as the foundational encoder
f[39]. - Module Specialization:
- For each high-resource task
i(e.g., band gap prediction), initialize a copy of the backbone. - Choose a parameterization strategy:
- Full Module: Update all parameters of the backbone model via supervised learning on task
i. The resulting parametersθ_f^iconstitute the module [39]. - Adapter Module: Insert adapter layers between each layer of the frozen pre-trained backbone. Train only the adapter parameters
Δ_f^ion taski. These adapters form the module [39].
- Full Module: Update all parameters of the backbone model via supervised learning on task
- For each high-resource task
- Centralization: Store the trained module parameters (
θ_f^iorΔ_f^i) in the MoMa Hub, formally defined asℋ = {g_1, g_2, ..., g_N}[39].
Adaptive Module Composition (AMC) Protocol
Objective
To dynamically compose a task-specific model for a downstream material property prediction task by intelligently combining the most synergistic modules from the MoMa Hub [39].
Procedure
- Target Task Formulation: Prepare the labeled downstream dataset
𝒟 = {(x_1, y_1), (x_2, y_2), ..., (x_m, y_m)}, wherex_jrepresents a material structure andy_jits target property [39]. - Synergistic Module Selection: Execute the data-driven AMC algorithm:
- Module Fusion & Fine-tuning: Create a composed model by combining the selected modules according to the optimized weights. This composed model is subsequently fine-tuned on the target task data
𝒟for final adaptation [39].
Performance Analysis and Benchmarking
Quantitative Performance Metrics
Table 2: MoMa Performance on Diverse Material Property Prediction Tasks
| Evaluation Scenario | Key Metric | Performance Result | Comparative Baseline |
|---|---|---|---|
| Overall Accuracy | Average Improvement across 17 datasets | +14% (Average improvement) | Strongest pre-training baseline [39] |
| Task-Win Rate | Number of tasks where MoMa is superior | 16/17 tasks | All baselines [39] |
| Few-Shot Learning | Performance gain with limited data | Larger gains vs. standard fine-tuning | Conventional pre-train then fine-tune [39] |
| Continual Learning | Capability to incorporate new tasks/molecules | Successfully demonstrated | Shows framework scalability [39] |
Integration with DFT Validation Workflows
A critical application of MoMa is enhancing and validating high-throughput computational screening, particularly where Density Functional Theory (DFT) serves as the primary data source or validation tool.
Augmenting DFT+U Band Gap Predictions
Accurate prediction of electronic properties like band gaps in metal oxides remains a challenge for standard DFT. The DFT+U approach, which applies a Hubbard U correction to address electron self-interaction error, is commonly used but requires careful selection of U parameters for metal (Ud/f) and oxygen (Up) orbitals [13].
- MoMa Integration: A MoMa module can be specifically trained to predict the optimal (Up, Ud/f) pairs for a given metal oxide system. This module would be trained on a dataset of DFT+U calculations where the U parameters have been rigorously benchmarked against experimental band gaps and lattice parameters [13]. The workflow can be visualized as follows:
- Protocol: For a new metal oxide (e.g., ZrO₂), the specialized MoMa module predicts an optimal (Up, Ud/f) pair (e.g., (9 eV, 5 eV) for c-ZrO₂) [13]. This enables a more accurate and computationally efficient single-shot DFT+U calculation, bypassing extensive manual benchmarking.
Correcting DFT Formation Enthalpies
Systematic errors in DFT-predicted formation enthalpies can limit the reliability of phase stability calculations. Machine learning models can be trained to correct these errors [34].
- MoMa Integration: A dedicated module can be trained to predict the discrepancy
ΔH_fbetween DFT-calculated and experimentally measured formation enthalpies. The input features for this module should be structured to capture key chemical and structural effects, such as elemental concentrations, weighted atomic numbers, and interaction terms [34]. - Protocol: The formation enthalpy is calculated as:
H_f^corrected = H_f^DFT + ΔH_f^ML. The MoMa framework is particularly suited for this, as a specialized correction module can be adaptively composed into a workflow only when predicting phase stability, avoiding interference with other property predictions.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Implementing the MoMa Framework
| Category | Specific Tool / Resource | Function in Protocol |
|---|---|---|
| Software & Libraries | PyTorch / TensorFlow | Deep learning backend for module implementation and training [39]. |
| Matminer | Primary source for curating diverse material property datasets for module training [39]. | |
| Computational Methods | Density Functional Theory (DFT) | Generates high-fidelity training data and provides ground-truth validation for predictions [13] [34]. |
| DFT+U (Hubbard Correction) | Critical for obtaining accurate electronic properties (e.g., band gaps) of strongly correlated materials for training/data validation [13]. | |
| Model Architectures | Pre-trained Force Field Models (e.g., M3GNet) | Powerful backbone encoders (f) for initializing MoMa modules, providing a strong prior on material structures [39]. |
| Parameter-Efficient Adapters | Key technology for creating memory-efficient modules, enabling larger and more diverse MoMa Hubs [39]. |
Navigating Practical Hurdles: Data, Generalization, and Optimization Strategies
The integration of Density Functional Theory (DFT) and machine learning (ML) has revolutionized materials validation research, enabling the rapid prediction of properties from atomic structure. However, a significant bottleneck persists: the scarcity of high-quality, labeled data required to train robust ML models. Generating sufficient DFT data is computationally prohibitive, creating a critical barrier to discovery, particularly for novel material classes like organic semiconductors or complex nanomaterials. This challenge is acutely felt by researchers and drug development professionals who require reliable property predictions for targeted applications.
Domain adaptation and transfer learning have emerged as powerful techniques to conquer this data scarcity. These methodologies enable knowledge gleaned from data-rich source domains to be efficiently applied to data-sparse target domains. Within the context of DFT and ML integration, this can translate to leveraging large, public DFT databases to build models that perform accurately on proprietary, experimentally-measured, or more complex computational datasets. This article provides detailed application notes and experimental protocols for implementing these techniques, specifically framed to enhance the robustness and scope of material validation research.
Theoretical Foundations: Domain Adaptation and Transfer Learning
Core Concepts and Definitions
Understanding the distinction between domain adaptation and transfer learning is crucial for their correct application.
Transfer Learning: This is a broad umbrella term for a collection of machine learning techniques where a model developed for a task is reused as the starting point for a model on a second task. The connection is the utilization of the predictive function from the source task in the creation of the predictive function for the target task. The features, labels, and datasets can all be vastly different between the two tasks [40]. For example, a model trained to recognize objects in photographs can be adapted to identify objects in medical images [41].
Domain Adaptation: This is a specific subfield of transfer learning where the task remains the same, but the domain of the data changes. The core assumption is that the feature space and labels are consistent, but the underlying data distributions differ between the source and target domains [40]. In materials science, a common scenario is using a model trained on a large DFT-calculated dataset (source domain) and adapting it to make accurate predictions on a smaller set of experimental data (target domain), which may have a different statistical distribution due to experimental conditions or systematic errors.
The Problem of Dataset Redundancy and Overestimation
A critical, often-overlooked issue in applying ML to materials data is the inherent redundancy in large public databases like the Materials Project. These databases contain many highly similar materials due to historical "tinkering" in material design. When ML models are trained and tested on randomly split data from such redundant datasets, they achieve over-optimistic performance metrics that do not reflect their true predictive capability on novel, out-of-distribution (OOD) samples [12]. This overestimation masks the model's poor extrapolation performance, which is often the primary goal in materials discovery. Tools like MD-HIT have been developed to control this redundancy by ensuring no pair of samples in the training and test sets are overly similar, leading to a more realistic evaluation of model performance [12].
Application Notes: Techniques and Workflows
Protocol 1: Multi-Task Learning for Universal Property Prediction
Objective: To train a single, universal model that can accurately predict multiple material properties simultaneously from a unified descriptor, thereby improving data efficiency and model transferability.
Rationale: Traditional ML models are trained to predict a single property, which is inefficient with scarce data. Multi-task learning leverages correlations between different properties during training. The improved performance on one property can inform and enhance the learning of others, leading to a more robust and generalizable model [11].
Experimental Workflow:
- Descriptor Selection: Use a physically grounded, universal descriptor. The electronic charge density is ideal, as it uniquely determines all ground-state material properties according to the Hohenberg-Kohn theorem [11].
- Data Acquisition and Standardization: Curate a dataset from sources like the Materials Project. The 3D charge density data from CHGCAR files must be standardized, as their dimensions are material-dependent. Convert the 3D matrices into a series of 2D image snapshots along crystal axes using a well-designed interpolation scheme [11].
- Model Architecture: Employ a Multi-Scale Attention-Based 3D Convolutional Neural Network (MSA-3DCNN). This architecture is adept at recognizing the rich spatial information and subtle local features (e.g., electron accumulation near bonds) within the charge density data [11].
- Model Training:
- Input: Standardized charge density image snapshots.
- Output Vectors: Multiple output nodes, each corresponding to a different target property (e.g., formation energy, band gap, bulk modulus).
- Training Regime: Train the model to minimize a composite loss function that is the weighted sum of the losses for each individual property. This forces the shared layers of the network to learn a general representation that is informative for all tasks.
Key Insight: Research has demonstrated that a multi-task learning approach based solely on electronic charge density can predict eight different material properties with an average R² value of 0.78, outperforming single-task models which averaged an R² of 0.66. This confirms that multi-task learning significantly enhances prediction accuracy and transferability [11].
Diagram 1: A high-level workflow for a standard transfer learning or domain adaptation process in materials informatics.
Protocol 2: Active Machine Learning for Targeted Discovery
Objective: To efficiently explore a vast chemical space with minimal DFT calculations by iteratively guiding data acquisition towards promising candidates.
Rationale: Exhaustive high-throughput screening of all possible candidates in a large chemical space is computationally intractable. Active Machine Learning (AML) is a data-efficient strategy that uses a surrogate model to balance exploration of unknown regions of the chemical space with exploitation of currently known promising candidates [42].
Experimental Workflow (for Organic Semiconductor Discovery):
- Define Search Space: Establish a set of "morphing operations" based on molecular fragments of known organic semiconductors (e.g., Acenes, Thienoacenes). These operations (e.g., ring annelation, linker addition) allow the generative exploration of an unlimited, chemically sensible search space [42].
- Select Fitness Descriptors: Choose computationally efficient DFT descriptors that are proxies for target properties. For OSC discovery, key descriptors are:
- Charge Injection Efficiency (ϵalign): The energy difference between the HOMO level and the work function of a standard electrode (e.g., Gold).
- Intramolecular Reorganization Energy (λ): A measure of the charge carrier mobility, with lower values indicating higher mobility [42].
- Iterative AML Cycle:
- Step 1 (Initialization): Perform DFT calculations for a small, random set of initial molecules.
- Step 2 (Surrogate Modeling): Train a surrogate model (e.g., Gaussian Process Regression - GPR) on the acquired data to predict the descriptors and, crucially, its own prediction uncertainty for any candidate.
- Step 3 (Candidate Selection & Query): Select the next candidates for DFT calculation by prioritizing those with high predicted fitness (exploitation) and/or high prediction uncertainty (exploration).
- Step 4 (Model Update): Add the new data points to the training set and update the surrogate model.
- Step 5 (Termination): Repeat steps 2-4 until a performance target is met or the computational budget is exhausted.
Key Insight: This AML approach has been shown to rapidly identify known high-performance OSCs and novel candidates with superior charge conduction properties, drastically outperforming conventional computational funneling methods [42].
Quantitative Performance of ML Techniques
Table 1: Summary of ML Model Performance for Material Property Prediction
| ML Technique | Application Context | Key Descriptor | Reported Performance | Key Challenge Addressed |
|---|---|---|---|---|
| Multi-Task Learning [11] | Predicting 8 material properties | Electronic Charge Density | Avg. R² = 0.78 (Multi-Task) vs. 0.66 (Single-Task) | Improves accuracy & transferability by learning correlated tasks. |
| Active ML [42] | Discovering organic semiconductors | HOMO/LUMO levels, Reorganization Energy | Rapid identification of superior candidates vs. conventional screening. | Efficiently navigates vast chemical spaces with minimal data. |
| Random Forest/Extra Trees [43] | Predicting reorganization energy of organic molecules | Molecular descriptors from structure | Best performance among 22 tested regression models. | Provides accurate predictions for complex quantum mechanical properties. |
| Standard Model (with Redundancy) [12] | Formation energy prediction | Composition/Structure | Overestimated performance with random dataset splits. | Highlights the risk of overfitting to redundant data. |
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Computational Tools for DFT-ML Integration
| Tool / Resource | Type | Function in Research | Relevance to Data Scarcity |
|---|---|---|---|
| Materials Project [11] [12] | Database | Source of high-throughput DFT data for thousands of materials (formation energy, band structure, etc.). | Primary source for large-scale source domain data for pre-training. |
| VASP (Vienna Ab initio Simulation Package) [11] [44] | Software | Performs quantum mechanical DFT calculations to compute material properties from first principles. | Generates high-fidelity target domain data, but is computationally expensive. |
| MD-HIT [12] | Algorithm | Controls redundancy in material datasets to prevent overestimated performance and improve OOD generalization. | Critical for creating realistic training/test splits and evaluating true model capability. |
| Gaussian 09 [43] | Software | Quantum chemistry package for calculating molecular properties, such as reorganization energy for organic molecules. | Generates target data for molecular/organic systems. |
| Python ML Stack (e.g., PyTorch, TensorFlow) [41] | Library/Framework | Provides flexible environments for building and training deep learning models, including transfer learning. | Enables implementation of fine-tuning, feature extraction, and domain adaptation algorithms. |
| Domain-Adversarial Neural Networks (DANN) [41] | Framework | A specific ML architecture for unsupervised domain adaptation that learns domain-invariant features. | Directly addresses domain shift between source (DFT) and target (experimental) data. |
Advanced Visualization: The Active Learning Cycle
The following diagram details the iterative workflow of an Active Machine Learning campaign, a powerful method for overcoming data scarcity in exploration.
Diagram 2: The iterative cycle of Active Machine Learning (AML) for materials discovery.
Domain adaptation and transfer learning are not merely auxiliary techniques but are foundational to the future of data-driven materials validation research. By providing structured protocols for multi-task learning and active machine learning, this article offers a pragmatic roadmap for researchers to overcome the critical challenge of data scarcity. The integration of these advanced ML strategies with the physical rigor of DFT, as demonstrated in the application notes, enables a more efficient and predictive framework. This empowers scientists to accelerate the discovery and validation of next-generation materials, from high-performance organic semiconductors to complex nanomaterials, even when starting from severely limited experimental or computational data.
The integration of Density Functional Theory (DFT) and machine learning (ML) has ushered in a new paradigm for materials discovery and validation research. A central tenet of this approach is the development of models with strong generalization capability, enabling accurate predictions for novel, unseen materials systems. Standard practice often involves evaluating model performance using random splits of a dataset, which assesses in-distribution (ID) generalization but fails to test a model's ability to handle realistic scenarios involving new chemical spaces or structural symmetries not present in the training data. This critical challenge is the problem of out-of-distribution (OOD) generalization [45].
Heuristic, human-defined OOD tests—such as leaving out all materials containing a specific element—are common but can be misleading. Recent evidence suggests that many such tests do not constitute true extrapolation; instead, most test data often reside within regions well-covered by the training data's representation space. This leads to overoptimistic conclusions about a model's generalizability and the benefits of neural scaling laws, as performance plummets on the minority of tasks that represent genuinely challenging OOD scenarios [45]. This application note details protocols and solutions for rigorously addressing the OOD generalization problem within DFT-ML pipelines for material validation.
Quantitative Assessment of OOD Generalization Gaps
Systematic evaluations across large materials databases reveal significant performance disparities between ID and OOD settings, and between different types of OOD tasks. The following tables summarize key quantitative findings.
Table 1: Performance Comparison of ID vs. OOD Tasks on Formation Energy Prediction (Materials Project Dataset) [45]
| Evaluation Setting | Best Model | Typical R² Score | Typical MAE (eV/atom) |
|---|---|---|---|
| In-Distribution (Random Split) | ALIGNN | > 0.98 | < 0.05 |
| OOD (Leave-One-Element-Out) | ALIGNN | 85% of tasks > 0.95 | Varies significantly |
| OOD (Challenging Elements: H, F, O) | ALIGNN | Poor (Negative possible) | High, with systematic bias |
Table 2: Analysis of OOD Generalization Across Different Splitting Strategies [45]
| OOD Splitting Criterion | Example | Physicochemical Meaning | Generalization Challenge |
|---|---|---|---|
| Leave-One-Element-Out | Train without any H-containing materials | Chemical dissimilarity, bonding uniqueness | High for H, F, O; Low for most metals |
| Leave-One-Group-Out | Train without any Group 15 (Pnictogen) materials | Valence electron configuration | Varies by group |
| Leave-One-Crystal-System-Out | Train without any Cubic materials | Structural symmetry and periodicity | Dependent on property-structure relationship |
| Space/Point Group | Train without a specific space group | Detailed symmetry operations | Can be high for unique symmetries |
The data shows that while models like graph neural networks (e.g., ALIGNN) and boosted trees (e.g., XGBoost) demonstrate surprising robustness on many heuristic OOD tasks, they fail catastrophically on specific challenges, such as predicting the formation energy of materials containing hydrogen (H), oxygen (O), or fluorine (F). For these tasks, the R² score can drop drastically, and errors are characterized by systematic biases rather than random noise, indicating a fundamental failure to capture the underlying physics [45].
Experimental Protocols for OOD Evaluation in Materials ML
To move beyond heuristic splits, researchers should adopt the following rigorous experimental protocols.
Protocol 1: Creating Physically Meaningful OOD Splits
Objective: To partition a materials dataset into training and test sets such that the test set represents a genuine distribution shift based on physicochemical principles.
- Define the Splitting Attribute: Select a physically meaningful attribute upon which to base the split. Common choices include:
- Elemental Identity: Leave out all compounds containing a specific element (e.g.,
Element == H). - Crystal System: Leave out all crystals belonging to a specific system (e.g.,
Crystal_System == 'Trigonal'). - Space Group: Leave out all materials with a specific space group number.
- Elemental Identity: Leave out all compounds containing a specific element (e.g.,
- Filter and Validate Splits:
- Apply the defined rule to partition the data. For example, to create a "Leave-One-Element-Out" split for Hydrogen, assign all entries where the chemical formula contains
Hto the test set, and all other entries to the training set. - Ensure the test set is of sufficient size (e.g., >200 samples) for statistically meaningful evaluation [45].
- For multi-attribute splits (e.g., leaving out a specific crystal system and element), ensure the logical criteria are consistently applied.
- Apply the defined rule to partition the data. For example, to create a "Leave-One-Element-Out" split for Hydrogen, assign all entries where the chemical formula contains
- Data Preprocessing: For the training set, standardize features (e.g., descriptors, fingerprints) by subtracting the mean and dividing by the standard deviation. Crucially, apply the scaler fitted on the training data to the test data to avoid data leakage.
Protocol 2: Model Agnostic OOD Benchmarking
Objective: To evaluate the OOD generalization performance of a diverse set of ML models, from simple to complex, on the defined splits.
- Model Selection: Train and evaluate a portfolio of models on the same OOD splits. This should include:
- Descriptor-Based Models: Random Forest (RF) and XGBoost (XGB) using human-engineered fingerprints (e.g., Matminer descriptors) [45].
- Graph Neural Networks (GNNs): Models like ALIGNN that operate directly on the crystal graph [45] [46].
- Neural Network Potentials: Models like Deep Potential (DP) that use alternative representations [47].
- Large-Scale Models: Transformer-based or other foundational models for materials science [45].
- Training and Evaluation:
- For each model and OOD split, train the model exclusively on the training partition.
- Evaluate on the held-out test set using metrics relevant to the prediction task (e.g., Mean Absolute Error (MAE), Root Mean Squared Error (RMSE), and Coefficient of Determination (R²)).
- Perform a comparative analysis of results as shown in Table 1.
Protocol 3: Diagnosing Failure Modes with Representation Analysis
Objective: To understand why a model fails on a particular OOD task by analyzing the learned materials representation space.
- Generate Representations: For a given model (e.g., a GNN), extract the latent vector representation (the output of the final hidden layer) for all materials in both the training and OOD test sets.
- Dimensionality Reduction: Use techniques like Principal Component Analysis (PCA) or t-SNE to project the high-dimensional latent vectors into a 2D or 3D space for visualization [47].
- Analyze Domain Coverage: Plot the projected representations, color-coding points by their dataset origin (training vs. test). A well-performing OOD task will typically show the test data residing within or near the domain of the training data. A failing task will often show the test data in a region of representation space that is poorly covered by the training set, indicating a true extrapolation [45].
- Identify Bias Sources (SHAP Analysis): For tasks with poor performance, train a secondary model (e.g., a shallow decision tree) to predict the error of the primary model. Use SHAP (SHapley Additive exPlanations) analysis on this meta-model to determine whether compositional features (e.g., electronegativity) or structural features (e.g., coordination number) are the primary drivers of the prediction error [45].
Diagram 1: A workflow for systematic evaluation and iterative improvement of OOD generalization.
The Scientist's Toolkit: Key Research Reagent Solutions
The following table catalogues essential computational "reagents" – databases, tools, and descriptors – critical for conducting rigorous OOD generalization research in computational materials science.
Table 3: Essential Research Reagent Solutions for OOD-Generalizable DFT-ML
| Research Reagent | Type | Function in OOD Research | Reference/Availability |
|---|---|---|---|
| JARVIS/MP/OQMD Databases | Data | Provides large-scale, ab-initio derived datasets for creating diverse OOD benchmarks across chemistry and structure. | [45] [46] |
| Electronic Charge Density (ρ) | Descriptor | A physically rigorous, universal descriptor from DFT; serves as a powerful input for predicting multiple properties, enhancing transferability. | [3] [11] |
| ALIGNN Model | Software/Tool | A graph neural network that models crystal structures; a state-of-the-art benchmark for OOD performance on materials property prediction. | [45] [46] |
| Matminer Fingerprints | Descriptor | Human-engineered, compositional, and structural features for training descriptor-based models (RF, XGB) as baselines for OOD tasks. | [45] |
| SHAP Analysis | Diagnostic Tool | Explains model predictions/errors, identifying if failure is due to chemical or structural biases in OOD data. | [45] |
| DP-GEN Framework | Software/Tool | Enables active learning and generation of neural network potentials, useful for building robust models via iterative data augmentation. | [47] |
Advanced Strategy: Universal Descriptors and Multi-Task Learning
For truly robust OOD generalization, moving beyond structure-based fingerprints to more fundamental physical descriptors is a promising path. The electronic charge density (ρ(r)), a core concept in DFT, provides a complete description of the ground state of a material system. Using ρ(r) as a unified input descriptor has been shown to enable accurate prediction of multiple properties (e.g., density of states, energy, forces) within a single model framework [3] [11].
This approach demonstrates outstanding multi-task learning capability, where prediction accuracy for individual properties often improves when more target properties are incorporated during training. This suggests that learning the underlying electronic structure simultaneously across multiple tasks forces the model to develop a more general and robust internal representation, which directly enhances OOD generalization and transferability across the materials space [11].
Diagram 2: A universal ML framework using electronic charge density as a single, physically grounded descriptor for multi-property prediction, which enhances OOD generalization.
In conclusion, addressing the OOD generalization problem requires a deliberate shift from convenient but flawed evaluation methods to a rigorous, physics-informed protocol. By adopting the defined splitting strategies, multi-model benchmarking, diagnostic analyses, and leveraging universal descriptors like the electronic charge density, researchers can build more trustworthy and robust ML models that truly accelerate materials validation and discovery.
Machine learning force fields (MLFFs) have emerged as powerful tools for atomistic simulations, offering near-quantum mechanical accuracy at a fraction of the computational cost of traditional ab-initio methods. However, their widespread adoption in materials validation research is often hampered by a critical challenge: physical robustness. MLFFs can produce unstable simulations that irreversibly drift into unphysical regions of phase space, leading to inaccurate property estimation or complete simulation collapse [48]. This application note, framed within a broader thesis on integrating density functional theory (DFT) and machine learning, details established and emerging protocols to ensure the stability and reliability of MLFFs for rigorous scientific and industrial applications, including drug development.
Core Challenges and Solutions in MLFF Training
A purely data-driven approach to MLFF development often encounters specific pitfalls that compromise robustness. The table below summarizes the primary challenges and corresponding strategic solutions.
Table 1: Key Challenges and Strategic Solutions for Robust MLFF Development
| Challenge | Impact on Simulation | Proposed Solution | Key Benefit |
|---|---|---|---|
| Inadequate Short-Range Repulsion [49] [50] | Unphysical atomic clustering and simulation breakdown due to poor extrapolation at short interatomic distances. | Hybrid MLFF frameworks incorporating empirical repulsive potentials (e.g., ZBL). | Built-in physical constraints prevent unphysical states; enhances training efficiency. |
| Insufficient Phase Space Sampling [28] [48] | Poor generalization and instability when simulation samples configurations absent from training data. | Active learning & stability-aware training (e.g., StABlE Training) that iteratively identifies and corrects unstable regions. | Improves model robustness and accuracy for long-time-scale dynamics. |
| Dataset Redundancy [12] | Overestimated model performance and poor out-of-distribution generalization. | Redundancy control algorithms (e.g., MD-HIT) to create balanced, non-redundant training sets. | Provides a more realistic evaluation of model performance and extrapolation capability. |
| Treatment of Complex Atomic Environments [28] | Higher model errors for atoms in different chemical environments (e.g., varying oxidation states). | Splitting atomic species into subtypes (e.g., O1, O2 for bulk/surface oxygen) during training. | Improves descriptive accuracy for heterogeneous systems. |
Detailed Experimental Protocols
Protocol 1: Implementing a Hybrid MLFF with Empirical Repulsion
This protocol leverages the hybrid NEP-ZBL framework to prevent unphysical atomic clustering in systems like solid electrolytes (LLZO) [49] [50].
Initial MLFF Training:
- Data Generation: Perform short ab-initio molecular dynamics (AIMD) simulations using VASP to generate an initial training set. Ensure electronic minimization is well-converged (check convergence of forces) and avoid using
MAXMIX > 0to prevent non-converged electronic structures [28]. - System Setup: Use
ISYM=0to turn off symmetry. For MD in the NpT ensemble, setENCUTat least 30% higher than for fixed-volume calculations [28]. - MLFF Training: Train a baseline Neuroevolution Potential (NEP) or other MLFF on the generated dataset.
- Data Generation: Perform short ab-initio molecular dynamics (AIMD) simulations using VASP to generate an initial training set. Ensure electronic minimization is well-converged (check convergence of forces) and avoid using
Integration of ZBL Potential:
- The total energy in the hybrid framework is computed as:
E_total = E_NEP + f(r) * E_ZBL, wheref(r)is a switching function that activates the ZBL potential at short distances [50]. - The ZBL potential for an atom pair i, j is:
U_ZBL(rij) = (1/(4πϵ0)) * (Zi * Zj * e² / rij) * φ(rij/a) * fc(rij)where Z is atomic number, e is elementary charge, r_ij is interatomic distance, a is a screening length, and φ is the universal screening function [49].
- The total energy in the hybrid framework is computed as:
Performance Validation:
- Dimer Curves: Calculate the potential energy curve for a dimer (e.g., Li-Li) to confirm the model exhibits a physically correct repulsive wall at short distances [50].
- Long-Time MD: Run extended molecular dynamics simulations (e.g., >1 ns) and monitor for unphysical clustering, checking radial distribution functions (RDFs) for stability [49].
- Property Prediction: Validate the model's ability to accurately reproduce key material properties, such as ionic conductivity via mean-squared displacement (MSD) analysis and phase transitions [50].
Protocol 2: Stability-Aware Training with Differentiable Boltzmann Estimators
The StABlE Training protocol uses system observables to correct instabilities without additional DFT calculations [48].
Initial Model and Data:
- Start with an MLFF pre-trained on a dataset of reference quantum-mechanical energies and forces.
- Have reference system observables (e.g., Radial Distribution Functions, virial stress) available for the target system.
Iterative Stability-Aware Training:
- Parallel Exploration: Run a large number (e.g., 100s) of independent MD simulations in parallel using the current MLFF.
- Instability Identification: Monitor these simulations to identify configurations where the model becomes unstable or samples unphysical states.
- Observable-Based Correction: Instead of performing new DFT calculations, correct the MLFF's behavior in these unstable regions by comparing simulation-derived observables against reference observables. This is achieved by minimizing a loss function through the Boltzmann Estimator, which enables gradient-based learning without backpropagating through the entire MD simulation [48].
- Model Update: Refine the MLFF parameters using this combined supervision from reference QM data and system observables.
Convergence and Validation:
- Iterate until simulation stability metrics (e.g., mean time before failure) converge.
- Validate the final model on held-out observables and its ability to perform stable, long-time-scale MD simulations at different temperatures.
Diagram 1: StABlE Training Workflow for MLFF Robustness.
This section lists key software, algorithms, and data handling techniques essential for developing robust MLFFs.
Table 2: Essential Tools for Robust MLFF Development
| Tool Name / Category | Type | Primary Function in MLFF Workflow |
|---|---|---|
| VASP [28] | Software Package | First-principles electronic structure calculations using DFT to generate reference training data (energies, forces, stresses). |
| Neuroevolution Potential (NEP) [49] [50] | Machine Learning Potential Framework | A highly efficient MLFF framework used as a base for hybrid models (e.g., NEP-ZBL). |
| Deep Potential (DP) [47] | Machine Learning Potential Framework | A neural network potential framework, often used with a generator (DP-GEN) for active learning. |
| ZBL Potential [49] [50] | Empirical Potential | Provides a physically accurate description of short-range repulsive nuclear interactions; integrated into hybrid MLFFs. |
| StABlE Training [48] | Training Algorithm | A multi-modal training procedure that uses system observables and QM data to improve simulation stability. |
| Boltzmann Estimator [48] | Computational Method | Enables gradient-based learning through MD simulations, allowing for efficient correction of instabilities. |
| MD-HIT [12] | Data Curation Algorithm | Controls redundancy in material datasets to prevent over-optimistic performance evaluation and improve generalizability. |
| DP-GEN [47] | Active Learning Framework | Automates the process of generating training data and building MLFFs through iterative sampling of configurations. |
Diagram 2: A Unified Workflow for Robust MLFF Development.
Application Note: Integrated DFT-ML Workflow for Accelerated Materials Discovery
The integration of Density Functional Theory (DFT) with Machine Learning (ML) has emerged as a transformative paradigm in computational materials science, particularly for the validation and discovery of novel materials. This approach effectively bridges the gap between the high accuracy but computational expense of first-principles calculations and the rapid screening capabilities of data-driven models. Standard DFT calculations often fail to correctly predict electronic properties like band gaps in complex systems such as metal oxides due to delocalization or self-interaction error [13]. While advanced DFT+U or hybrid functional methods can improve accuracy, they come with significant computational overhead [13]. Machine learning models, once trained on high-quality DFT or experimental data, can predict key material properties with orders of magnitude greater efficiency, allowing for rapid screening of vast chemical spaces [6]. This application note details a streamlined workflow that synergistically combines these methodologies for efficient material property prediction and validation.
Core Integrated DFT-ML Workflow
The following diagram illustrates the optimized, cyclical workflow for integrating DFT and ML in materials research, detailing the sequential stages from initial data generation to final model deployment and validation.
Figure 1: Integrated DFT-ML workflow for materials research. This cyclical process enables continuous improvement through active learning.
Workflow Stage Protocols
Stage 1: Data Generation via High-Throughput DFT
The foundation of any robust ML model is a high-quality, diverse dataset. This stage focuses on generating reference data using high-throughput DFT calculations.
Protocol 1.1: DFT+U for Metal Oxides
- Objective: Accurately predict band gaps and lattice parameters for strongly correlated systems.
- Methodology:
- Validation: Compare predicted band gaps and lattice parameters with experimental values.
- Output: Benchmark dataset of electronic structures for metal oxides.
Protocol 1.2: Formation Enthalpy Calculations
- Objective: Compute accurate formation enthalpies for phase stability assessment.
- Methodology:
- Output: Formation enthalpy dataset for binary and ternary alloys.
Stage 2: Feature Engineering and Data Preparation
Transform raw DFT outputs into meaningful descriptors for ML model training.
Protocol 2.1: Material Descriptor Generation
- Input: DFT-calculated structural and electronic properties.
- Feature Types:
- Elemental concentrations and weighted atomic numbers [5].
- Structural descriptors (lattice parameters, symmetry information).
- Electronic descriptors (band structure moments, density of states features).
- Thermodynamic descriptors (formation energy, phonon frequencies).
- Preprocessing: Standardize and normalize features to ensure uniform scales [51].
Protocol 2.2: Data Quality Control
- Objective: Ensure dataset reliability for model training.
- Methods:
Stage 3: Machine Learning Model Training
Develop predictive models using the curated DFT dataset.
Protocol 3.1: Model Selection and Training
- Algorithm Options:
- Training Protocol:
Protocol 3.2: Transfer Learning Implementation
- Objective: Enhance performance on small datasets.
- Strategies:
- Validation: Test on out-of-distribution datasets to assess generalizability [51].
Stage 4: Model Validation and DFT Verification
Ensure model reliability through rigorous validation and first-principles verification.
Protocol 4.1: Model Performance Assessment
- Metrics: Mean Absolute Error (MAE), Root Mean Square Error (RMSE), R$^2$ score.
- Methods:
- Leave-one-out cross-validation for small datasets [5].
- Train-test splits with stratified sampling.
- Comparison against baseline DFT calculations.
Protocol 4.2: First-Principles Validation
- Objective: Verify ML predictions using DFT calculations.
- Methodology:
- Select top candidates identified by ML screening [6].
- Perform full DFT relaxation and property calculation.
- Compare ML-predicted vs. DFT-calculated properties.
- Iterate workflow if discrepancies exceed acceptable thresholds.
Performance Benchmarks and Validation
The following table summarizes quantitative performance benchmarks for integrated DFT-ML workflows across various material systems and applications.
Table 1: Performance Benchmarks for DFT-ML Workflows in Materials Research
| Material System | Target Property | ML Model | Performance Metrics | Reference |
|---|---|---|---|---|
| Metal Oxides (TiO$2$, ZnO, CeO$2$) | Band Gap, Lattice Parameters | Supervised Regression | Closely reproduces DFT+U results at fraction of computational cost | [13] |
| Alkali-Metal-Ion Batteries | Average Voltage | Deep Neural Network (DNN) | Strong predictive performance corroborated by DFT calculations | [6] |
| Binary/Ternary Alloys | Formation Enthalpy | Neural Network (MLP) | Significant enhancement of predictive accuracy for phase stability | [5] |
| Diverse Material Classes | Multiple Properties | Transfer Learning with GNNs | Outperforms models trained from scratch on small datasets | [51] |
| Organic Dyes (DSSCs) | Power Conversion Efficiency | Two-Stage ML with DFT | Identification of novel high-performance dye candidates | [54] |
Research Reagent Solutions
The following table details essential computational tools and resources for implementing the integrated DFT-ML workflow.
Table 2: Essential Research Reagents for DFT-ML Integration
| Reagent/Tool | Type | Function | Application Example |
|---|---|---|---|
| VASP | DFT Software | First-principles electronic structure calculations | High-throughput screening of metal oxide properties [13] |
| EMTO-CPA | DFT Software | Total energy calculations for disordered alloys | Formation enthalpy calculations for phase stability [5] |
| PyTorch/TensorFlow | ML Framework | Deep neural network implementation | Voltage prediction for battery materials [6] |
| RDKit | Cheminformatics | Molecular descriptor generation and manipulation | Fragmenting and recombining dye building blocks [54] |
| Materials Project | Database | Repository of calculated material properties | Source of training data for various property prediction models [6] |
Advanced Protocol: Transfer Learning for Limited Data Scenarios
Rationale and Workflow
Many materials science applications face data scarcity challenges, particularly for experimental properties or complex synthesized materials. Transfer learning (TL) addresses this limitation by leveraging knowledge from large datasets to enhance performance on smaller target datasets. The following diagram illustrates the transfer learning workflow for material property prediction.
Figure 2: Transfer learning workflow for material property prediction with limited data.
Implementation Protocol
Protocol 5.1: Multi-Stage Pre-training
- Objective: Leverage large materials datasets to learn general feature representations.
- Source Datasets: Formation energy (132,752 materials), DFT band gap (106,113 structures), average shear modulus (10,987 materials) [51].
- Model Architecture: Graph Neural Networks (GNNs) that represent materials as graphs with atoms as nodes and bonds as edges [51].
- Output: Pre-trained model with learned chemical and structural representations.
Protocol 5.2: Fine-tuning Strategies
- Strategy 1: Unfreeze all layers for complete model adaptation [51].
- Strategy 2: Add new prediction head while keeping base layers frozen [51].
- Strategy 3: Unfreeze only the last layer for minimal adaptation [51].
- Strategy 4: Unfreeze selective layers based on domain similarity assessment [51].
- Validation: Test on completely different datasets (e.g., 2D material band gaps) to assess generalizability [51].
Performance Validation
Studies demonstrate that transfer learning models consistently outperform models trained from scratch on small target datasets [51]. Multi-property training (simultaneous pre-training on multiple properties) further enhances performance and generalizability compared to conventional pair-wise transfer learning approaches [51].
Benchmarks and Reality Checks: Rigorously Validating ML Models Against DFT and Experiment
The integration of Density Functional Theory (DFT) and machine learning (ML) has revolutionized atomistic simulations, enabling the study of complex molecules, materials, and interfaces with quantum-mechanical accuracy at a fraction of the computational cost. Central to this paradigm are Machine Learning Force Fields (MLFFs), which are trained on quantum-mechanical energies and forces to predict potential energy surfaces. Despite their remarkable success, the development of generally useful MLFFs is challenged by issues of reliable transferability, reactivity, and scalability, particularly for organic chemistry and softer materials [55]. The inherent complexity and occasional opacity of ML models necessitate rigorous, independent validation to ensure their predictions are reliable for safety-critical applications, such as drug development and material design [56].
The "Crash testing machine learning force fields for molecules, materials, and interfaces" initiative, known as the TEA Challenge 2023, was established to provide a platform for such rigorous testing. It gathered leading MLFF developers to evaluate their models on standardized, diverse datasets under identical conditions. This challenge simulated realistic application conditions where the ground truth is often unknown, thereby highlighting potential issues practitioners might encounter. The subsequent analysis provided an unprecedented comparative look at the performance of various MLFF architectures, offering invaluable insights into their accuracy, stability, and limitations [55] [57]. This application note synthesizes the key findings and methodologies from the TEA Challenge, providing a framework for the validation of MLFFs within material validation research.
TEA Challenge 2023: Experimental Design and Evaluation Framework
Challenge Architecture and Participating Models
The TEA Challenge 2023 was designed to impartially assess the capabilities of modern MLFFs across a spectrum of realistic scenarios. The organizers provided training datasets with limited information about data generation details to prevent unilateral data extension. Participating developers then trained their models, making independent choices regarding model size, accuracy, and computational efficiency trade-offs. The final submitted models represented a range of approaches, from lighter kernel regression models to heavier neural networks [55].
The following MLFF architectures participated in the challenge, representing the state of the art as of October 2023:
After model submission, the organizers conducted independent molecular dynamics (MD) simulations using the final MLFF models under identical conditions on the same High Performance Cluster (HPC). This phase was crucial for testing the models' performance in a realistic application setting where the ground truth is unknown [55].
Benchmark Datasets and Challenge Categories
The challenge was structured around four distinct datasets, each designed to probe specific capabilities and limitations of MLFFs [55]:
- Challenge I: Reproducing Potential Energy Surfaces. This challenge evaluated the core ability of MLFFs to accurately reproduce quantum-mechanical potential energy surfaces for molecular systems.
- Challenge II: Handling Incomplete Reference Data. This tested model robustness and generalization when training data is sparse or non-exhaustive.
- Challenge III: Managing Multi-component Systems. This assessed performance on systems containing multiple chemical elements or components.
- Challenge IV: Modeling Complex Periodic Structures. This focused on the ability to simulate extended periodic materials, a key requirement for materials science applications.
Key Performance Metrics and Quantitative Results
The evaluation of MLFFs in the TEA Challenge was based on three principal criteria: the accuracy of energy and force predictions compared to reference DFT calculations, the stability of models during extended molecular dynamics simulations, and the computational efficiency required to produce dynamics.
Table 1: Summary of MLFF Model Performance in the TEA Challenge 2023
| MLFF Model | Reported Force RMSE (kcal mol⁻¹ Å⁻¹) | MD Stability Performance | Noted Strengths and Limitations |
|---|---|---|---|
| MACE | Sub-kcal/mol accuracy achieved by participating models [55] | Stable across diverse applications [57] | Weak dependency on architecture when problem is within scope; performance heavily dataset-dependent [57] |
| SO3krates | Sub-kcal/mol accuracy achieved by participating models [55] | Stable across diverse applications [57] | Weak dependency on architecture when problem is within scope; performance heavily dataset-dependent [57] |
| sGDML | Sub-kcal/mol accuracy achieved by participating models [55] | Stable across diverse applications [57] | Weak dependency on architecture when problem is within scope; performance heavily dataset-dependent [57] |
| SOAP/GAP | Sub-kcal/mol accuracy achieved by participating models [55] | Stable across diverse applications [57] | Weak dependency on architecture when problem is within scope; performance heavily dataset-dependent [57] |
| FCHL19* | Sub-kcal/mol accuracy achieved by participating models [55] | Stable across diverse applications [57] | Weak dependency on architecture when problem is within scope; performance heavily dataset-dependent [57] |
A central finding was that all participating models demonstrated the capacity to achieve root mean squared errors in forces within a fraction of one kcal (mol⁻¹ Å⁻¹) compared to reference quantum chemistry methods. Furthermore, when a problem falls within the scope of a given MLFF architecture, the resulting simulations exhibit a weak dependency on the specific architecture used. The emphasis for achieving reliable results should, therefore, be placed on developing complete, reliable, and representative training datasets [57].
^The specific quantitative force RMSE for each model is detailed in the primary source as being sub-kcal/mol, but exact comparative values for each model are not fully enumerated in the provided excerpts. The findings reflect the state of ML architectures as of October 2023.
Table 2: Computational Efficiency Benchmark of MLFFs
| Model Type | Parameter Count Range | Relative Computational Efficiency for MD |
|---|---|---|
| Kernel Regression Models | ~123,000 parameters [55] | High efficiency |
| Lighter Neural Networks | ~487,613 parameters [55] | Medium efficiency |
| Heavier Neural Networks | ~2,983,184 parameters [55] | Lower efficiency (but context-dependent) |
Critical Limitations and Failure Mode Analysis
Despite the overall promising performance, the TEA Challenge identified specific, critical limitations common to many MLFFs.
The Long-Range Interaction Challenge
A paramount finding was that long-range noncovalent interactions remain challenging for all evaluated MLFF models. This necessitates special caution in simulations of physical systems where such interactions are prominent, such as molecule-surface interfaces [57]. The failure to accurately capture these interactions can lead to quantitatively and qualitatively incorrect simulation results, potentially undermining the predictive power of the model for applications like drug binding or surface adsorption.
Data Dependency and Transferability
The challenge confirmed that the performance and reliability of MLFFs are profoundly dependent on the quality and representativeness of the training data. A model is only as good as the data it was trained on; incomplete or non-representative datasets lead to models that fail to generalize to new regions of chemical space [55]. This highlights a key difference from traditional DFT, where the functional is transferable across systems.
Silent Failures and Model Robustness
A significant risk identified in the use of MLFFs is the occurrence of "silent failures," where the model does not fail outright but produces seemingly reasonable yet ultimately incorrect results. Identifying such cases in large and complex systems requires deeper assessment than a mere comparison to reference ab initio calculations, as those calculations can be computationally prohibitive or even unfeasible [55]. This underscores the need for robust validation protocols beyond simple error metrics.
Application Notes: Protocols for MLFF Validation
Based on the TEA Challenge framework, the following protocols are recommended for researchers integrating MLFFs into material validation workflows.
Protocol for Cross-Model Validation
Purpose: To mitigate the risk of silent failures by leveraging the consensus and discrepancies between independent models. Procedure:
- Identify a critical system or property for validation where reference DFT is computationally expensive.
- Run identical MD simulations using at least two, but preferably more, independently developed MLFF architectures (e.g., MACE, sGDML).
- Compare key observables (e.g., radial distribution functions, diffusion coefficients, free energy profiles) obtained from the different simulations.
- Analysis: A consistent outcome across different architectures indicates a high likelihood of a correct result. When discrepancies arise, this is indicative of potential mistakes in one or all MLFF results, and the simulation results should be interrogated further with targeted DFT single-point calculations if possible [55].
Protocol for Stability and Long-Time Scale MD
Purpose: To evaluate the stability and physical correctness of an MLFF beyond short-time scale accuracy. Procedure:
- Perform extended molecular dynamics simulations (on the order of nanoseconds) under the relevant thermodynamic ensemble (NVT, NPT).
- Monitor the conservation of energy in microcanonical (NVE) simulations for isolated systems to check for numerical instability.
- For condensed-phase systems, track the evolution of structural properties (e.g., lattice parameters, volume) over time to ensure the model does not exhibit unphysical drift.
- Analysis: Stable conservation of energy and convergence of structural properties to physically realistic values are indicators of a robust model. This protocol tests the model in a regime far beyond its training data, probing its true predictive capability [55] [57].
Protocol for Testing Long-Range Interactions
Purpose: To specifically assess an MLFF's ability to handle noncovalent interactions at molecule-surface interfaces or in dispersed systems. Procedure:
- Construct a test system where long-range interactions are critical, such as a molecule adsorbed at a distance from a surface or a molecular crystal held by dispersion forces.
- Compute the binding or interaction energy curve as a function of distance using the MLFF.
- Compare this curve with a reference calculation using a high-level quantum mechanics method (e.g., DFT with a van der Waals-corrected functional).
- Analysis: Significant deviation from the reference curve, especially at intermediate to long range, indicates a limitation of the MLFF for simulating interfaces or soft matter systems. This protocol is essential before applying any MLFF to problems in catalysis or supramolecular assembly [57].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Computational Tools for MLFF Development and Validation
| Tool / Resource | Function in MLFF Workflow |
|---|---|
| DFT Codes (VASP, Gaussian, Quantum ESPRESSO) | Generates high-quality reference data (energies, forces) for training and testing MLFFs [55] [58]. |
| MLFF Software (MACE, SO3krates, sGDML) | Provides the core architectures and training algorithms for developing machine learning potentials. |
| Molecular Dynamics Engines (LAMMPS, ASE) | Performs the final molecular dynamics simulations using the trained MLFFs to compute observables. |
| Ab Initio Molecular Dynamics (AIMD) | Serves as a benchmark for short MD trajectories to validate the MLFF's dynamical correctness. |
| High-Performance Computing (HPC) Cluster | Provides the necessary computational resources for training models and running production MD simulations [55]. |
Workflow and Relationship Visualizations
TEA Challenge Validation Workflow
The following diagram illustrates the end-to-end workflow of the TEA Challenge, from data provision to final analysis, which serves as a template for rigorous MLFF validation.
MLFF Failure Mode and Mitigation Relationships
This diagram maps the primary failure modes identified in the TEA Challenge to their potential effects and recommended mitigation strategies, forming a basis for a failure mode and effects analysis (FMEA) in MLFF projects.
The TEA Challenge 2023 demonstrates that modern MLFFs have matured to a point where they can deliver highly accurate and efficient simulations for a wide range of systems. The finding that results show a weak dependency on the specific architecture, when the problem is within its scope, is empowering for practitioners, as it suggests the focus should shift from a search for a singular "best" model to the meticulous construction of training datasets and the implementation of rigorous validation protocols [57].
However, significant challenges remain, most notably the accurate description of long-range interactions. Future development must focus on incorporating physical principles for non-local interactions to improve performance at interfaces and in soft matter. Furthermore, the community would benefit from the standardization of benchmark challenges like the TEA Challenge to continuously monitor progress, identify new failure modes, and build trust in MLFFs for safety-critical material validation and drug development research. The protocols and analyses provided here offer a foundational framework for this ongoing endeavor.
The integration of Density Functional Theory (DFT) and Machine Learning (ML) has created a powerful paradigm for accelerating materials discovery and design [14] [59]. However, the predictive power of this hybrid approach ultimately depends on the physical validity of its outcomes. While DFT provides quantum mechanical insights and ML models offer rapid property predictions, their results require robust validation in a real-world context. This is where Molecular Dynamics (MD) simulations, coupled with the direct prediction of experimental observables, become indispensable. Moving beyond simple error metrics like root-mean-square deviation (RMSD), this protocol details how to use MD simulations as a validation bridge, ensuring that DFT- and ML-predicted structures and mechanisms are not only computationally plausible but also dynamically and thermodynamically consistent with empirical evidence [60]. This document provides application notes and detailed protocols for researchers to implement this critical validation step within their material and drug development workflows.
Comparative Analysis of MD Simulation Approaches
Selecting an appropriate MD simulation package and force field is a critical first step. Different software and parameter sets can produce congruent results for native-state dynamics but may diverge significantly when probing larger conformational changes, such as thermal unfolding [60]. The following table summarizes key characteristics of commonly used MD packages to guide initial selection.
Table 1: Comparison of Molecular Dynamics Software Packages
| Software Package | Example Force Fields | Typical Water Models | Key Considerations |
|---|---|---|---|
| AMBER [60] | ff99SB-ILDN [60] | TIP4P-EW [60] | Well-established for biomolecular simulations; uses periodic boundary conditions in a truncated octahedral box. |
| GROMACS [60] [61] | AMBER ff99SB-ILDN, CHARMM36 [60] | Varies (e.g., SPC, TIP4P) | High performance and efficiency; includes physical validation suites to check for unphysical behavior [61]. |
| NAMD [60] | CHARMM36 [60] [61] | Varies | Strengths in parallel scaling for large systems; often used with the CHARMM force field. |
| ilmm [60] | Levitt et al. [60] | Varies | — |
The choice of simulation strategy significantly impacts the conformational sampling achieved. As illustrated in the workflow below, a robust validation protocol involves multiple parallel simulations and rigorous checks to ensure physical reliability.
Detailed Experimental Protocol for MD Validation
System Setup and Simulation Parameters
This protocol, adapted from best practices in the literature [60], outlines the process for setting up and running MD simulations for a protein system, starting from an initial structure.
Initial System Preparation:
- Obtain Initial Coordinates: Use a structure sourced from the Protein Data Bank (PDB) or derived from DFT/ML modeling.
- Add Hydrogen Atoms: Model explicit hydrogen atoms onto the structure using the respective simulation package's tool (e.g., the
leapmodule in AMBER). - Solvation: Solvate the protein in an explicit water model (e.g., TIP4P-EW) within a periodic boundary box (e.g., a truncated octahedron) that extends at least 10 Å beyond any protein atom [60].
- Neutralization: Add ions to neutralize the system's net charge and to achieve a physiologically relevant ionic concentration.
Energy Minimization and Equilibration:
- Minimization: Perform a multi-stage energy minimization to remove bad contacts.
- Stage 1: Minimize solvent atoms with strong positional restraints (e.g., 100 kcal mol⁻¹) on protein atoms.
- Stage 2: Minimize solvent atoms and protein side chains, restraining the protein backbone.
- Stage 3: Perform a full minimization of the entire system without restraints [60].
- Heating: Gradually heat the system to the target temperature (e.g., 298 K) over 50-100 ps in the NVT ensemble, using a thermostat (e.g., Langevin) and weak restraints on the protein.
- Density Equilibration: Allow the system density to equilibrate by running a short simulation (e.g., 100 ps) in the NPT ensemble at the target temperature and pressure (e.g., 1 bar), using a barostat.
Production Simulation:
- Run Production MD: Conduct multiple independent simulation replicates (e.g., 3x 200 ns) in the NPT ensemble using a symplectic integrator (e.g., velocity Verlet or leap-frog) with a 2 fs time step [60].
- Employ Best Practices: Use best practice parameters for long-range electrostatics (e.g., Particle Mesh Ewald) and constrain bonds involving hydrogen atoms.
Physical Validity Testing
Before analyzing the results, it is crucial to verify the physical validity of the simulations. A two-fold approach is recommended [61].
Table 2: Essential Physical Validity Tests for MD Simulations
| Test Category | Description | Protocol | Interpretation |
|---|---|---|---|
| Integrator Validation [61] | Checks the correctness of the integration algorithm by verifying the relationship between energy fluctuations and time step. | Run multiple short simulations of the same system using different time steps (e.g., 1 fs and 2 fs). | The ratio of total energy fluctuations should scale with the square of the time step ratio. Significant deviation indicates a non-conservative integrator. |
| Ensemble Validation [61] | Checks if the simulation correctly samples the intended thermodynamic ensemble (e.g., NPT). | Compare the average simulated temperature and pressure with the intended set points. | Large, systematic deviations suggest incorrect ensemble sampling. |
| Ergodicity Check [61] | Assesses whether different degrees of freedom (e.g., solute vs. solvent) are in thermal equilibrium. | Compare the kinetic temperature of the solute with that of the solvent. | A significant difference (the "flying ice cube" effect) indicates non-ergodic behavior and unphysical energy transfer. |
These tests can be performed using open-source tools like the physical-validation Python library [61].
Predicting and Comparing Experimental Observables
The core of the validation process is the comparison of simulation-derived observables with experimental data. This moves beyond simple structural metrics to functional validation.
Key Observables and Comparison Methods:
Nuclear Magnetic Resonance (NMR) Data:
- Backbone NMR S² Order Parameters: Calculate from simulation trajectories using the equation: ( S^2 = \frac{1}{2} \langle 3\cos^2\theta - 1 \rangle ), where ( \theta ) is the angle of the N-H bond vector relative to its average orientation. Compare with experimentally derived values to validate backbone mobility [60].
- NMR Spin Relaxation: Use the Lipari-Szabo model-free approach or direct spectral density mapping from the simulation to predict relaxation rates (R₁, R₂) and the heteronuclear NOE [60].
Small-Angle X-Ray Scattering (SAXS):
- Compute the theoretical scattering profile, I(q), directly from the simulation trajectory by averaging over all frames.
- Compare the calculated profile with the experimental SAXS curve. A good fit indicates the simulation ensemble accurately represents the solution-state conformation and flexibility.
Protein Folding/Unfolding:
- Simulate at elevated temperatures (e.g., 498 K) to induce unfolding and monitor metrics like radius of gyration (Rg) and secondary structure content over time.
- Compare the unfolding pathways and the stability of structural elements with experimental data, if available. This is a stringent test of the force field's transferability [60].
The logical relationship between the DFT/ML model, MD simulation, and experimental validation is summarized below.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for MD Validation
| Tool / Resource | Type | Primary Function in Validation |
|---|---|---|
| AMBER [60] | MD Software Suite | A comprehensive package for simulating biomolecules, particularly known for its force fields and tools for analyzing NMR properties. |
| GROMACS [60] [61] | MD Software Suite | A high-performance engine for MD simulation, notable for including built-in physical validation tests to catch unphysical behavior. |
| CHARMM36 [60] | Force Field | An all-atom force field for proteins, nucleic acids, and lipids, providing parameters for simulations. |
| AMBER ff99SB-ILDN [60] | Force Field | A force field optimized for protein side chains and backbone dynamics, often used with the AMBER and GROMACS packages. |
| TIP4P-EW [60] | Water Model | An explicit water model designed for use with Ewald summation methods for electrostatics, improving the accuracy of water properties. |
| Physical-Validation Library [61] | Python Library | An open-source tool for testing the physical validity of MD simulations, including checks of integrators, ensembles, and ergodicity. |
| Particle Mesh Ewald (PME) [60] | Algorithm | A standard method for accurately treating long-range electrostatic interactions in periodic systems, critical for simulation realism. |
The integration of machine learning force fields (MLFFs) with density-functional theory (DFT) has emerged as a transformative approach for accelerating material validation research. MLFFs bridge the gap between computationally prohibitive quantum-mechanical calculations and efficient molecular dynamics (MD) simulations, enabling high-fidelity modeling of complex systems at extended time and length scales [62] [63]. This application note provides a structured comparison of prominent MLFF architectures—MACE, SO3krates, NequIP, and others—focusing on their theoretical foundations, performance characteristics, and practical implementation protocols for molecular and materials systems.
Theoretical Foundations of MLFF Architectures
Key Architectural Paradigms
Modern MLFF architectures can be broadly categorized into equivariant message-passing neural networks and kernel-based methods, each with distinct approaches to representing atomic interactions.
Table 1: Core Architectural Paradigms in Machine Learning Force Fields
| Architecture Type | Representative Models | Key Features | Mathematical Foundation |
|---|---|---|---|
| Equivariant Message-Passing Neural Networks | MACE, SO3krates, NequIP | E(3)-equivariance, directional information, many-body interactions | Spherical harmonics, tensor products, irreducible representations [62] [64] [65] |
| Kernel-Based Methods | sGDML, SOAP/GAP, FCHL19* | Kernel similarity measures, local/global descriptors | Kernel ridge regression, smooth overlap of atomic positions [66] |
Principles of Equivariance in MLFFs
Equivariance represents a fundamental physical symmetry that ensures model predictions transform correctly under rotational and translational operations. Formally, a function (f: X \rightarrow Y) is equivariant with respect to a group (G) if: [ {D}{Y}[g]f(x)=f({D}{X}[g]x)\quad \forall g\in G,\forall x\in X ] where (DX[g]) and (DY[g]) are representations of the group element (g) in the vector spaces (X) and (Y), respectively [64]. For MLFFs, E(3)-equivariance (covering rotations, reflections, and translations in 3D space) ensures that force vectors rotate appropriately with the system while the potential energy remains invariant [64].
The NequIP architecture implements equivariance through features comprised of tensors of different orders (scalars, vectors, and higher-order tensors) that form irreducible representations of the O(3) symmetry group [64]. Similarly, SO3krates employs a Euclidean self-attention mechanism that replaces expensive SO(3) convolutions with a filter on the relative orientation of atomic neighborhoods [62].
Figure 1: Computational workflow of equivariant MLFF architectures showing the transformation of atomic coordinates into predictions of energy and forces through equivariant operations.
Comparative Performance Analysis
TEA Challenge 2023 Benchmarking Results
The TEA Challenge 2023 provided a rigorous evaluation platform for modern MLFFs, testing models including MACE, SO3krates, sGDML, SOAP/GAP, and FCHL19* across diverse systems including biomolecules, molecule-surface interfaces, and periodic materials [66].
Table 2: Performance Comparison of MLFF Architectures from TEA Challenge 2023
| MLFF Architecture | Architecture Type | Key Strengths | Limitations | Representative Systems |
|---|---|---|---|---|
| MACE | Equivariant Message-Passing NN | Many-body interactions, high accuracy [66] | Computational cost | Molecules, materials [66] |
| SO3krates | Equivariant Transformer | Speed (30x faster), stability, exploration [62] | Implementation complexity | Peptides, nanostructures [62] |
| NequIP | Equivariant GNN | Exceptional data efficiency [64] | Training complexity | Small molecules, water, amorphous solids [64] |
| sGDML | Kernel-Based | Global descriptor accuracy [66] | Poor scaling to large systems | Small organic molecules [66] |
| SOAP/GAP | Kernel-Based | Local environment description [66] | Limited transferability | Materials, interfaces [66] |
Critical Performance Insights
The TEA Challenge revealed that when a problem falls within a model's scope, simulation results exhibit weak dependency on the specific architecture employed [66]. Instead, the completeness and representativeness of training datasets emerged as more critical factors than architectural nuances for many applications [66]. However, significant challenges remain in modeling long-range noncovalent interactions, particularly critical in molecule-surface interfaces where all tested MLFF models exhibited limitations [66].
SO3krates demonstrates a unique balance between stability and the emergence of new minimum-energy conformations beyond training data, which is crucial for realistic exploration tasks in biochemistry [62]. Meanwhile, NequIP achieves remarkable data efficiency, outperforming existing models with up to three orders of magnitude fewer training data, challenging the belief that deep neural networks require massive training sets [64].
Experimental Protocols and Methodologies
Standardized Benchmarking Workflow
Figure 2: Standardized workflow for benchmarking MLFF architectures, from system selection through validation against reference data.
Protocol 1: Biomolecular System Validation (Alaninetetrapeptide)
Purpose: To evaluate MLFF performance for flexible biomolecules with complex conformational landscapes [66].
Materials:
- Initial structure: Alanine tetrapeptide (AT) coordinates
- Reference data: DFT-level energies and forces
- Training set: 100-1000 configurations (for data efficiency testing)
- Simulation software: ASE, LAMMPS, or internal MD codes
Procedure:
- Reference Data Generation:
- Perform ab initio MD using DFT (PBE/DFT-D3) to sample diverse conformations
- Extract energies and forces for each snapshot
- Split data into training (80%), validation (10%), and test sets (10%)
Model Training:
- Configure MLFF with appropriate cutoffs (typically 4-6 Å)
- Set hyperparameters: learning rate (1e-3), batch size (5-10)
- Train until validation loss plateaus (typically 100-1000 epochs)
MD Simulations:
- Run 1-10 ns MD simulations at 300 K using trained MLFF
- Use NVT ensemble with Langevin thermostat
- Employ 0.5-1.0 fs time step
Analysis:
- Compare Ramachandran plots with DFT reference
- Analyze dihedral angle distributions
- Calculate root-mean-square deviation of key structural motifs
Validation Metrics: Potential energy distributions, force MAE (< 50 meV/Å desirable), conformational populations, free energy surfaces [66].
Protocol 2: Material System Validation (Methylammonium Lead Iodide Perovskite)
Purpose: To assess MLFF performance for periodic material systems with complex ionic interactions [66].
Materials:
- Crystal structure: MAPbI3 experimental coordinates
- DFT reference: PBEsol functional with SOC corrections
- Training data: 500-5000 configurations from ab initio MD
Procedure:
- Training Data Curation:
- Sample configurations from NPT ab initio MD at 300 K
- Include various octahedral tilting patterns
- Extract energies, forces, and stresses
Model Training:
- Enable periodic boundary conditions
- Use extended cutoffs (5-7 Å) for long-range interactions
- Include stress components in loss function
MD Simulations:
- Run 100-500 ps NPT simulations at ambient conditions
- Calculate phase stability and phase transition temperatures
Analysis:
- Compare radial distribution functions with DFT/experiment
- Analyze octahedral tilting dynamics
- Calculate phonon density of states
Validation Metrics: Lattice parameters (±1%), formation energies (±20 meV/atom), phase transition temperatures, phonon spectra [66].
Protocol 3: Interface System Validation (1,8-Naphthyridine/Graphene)
Purpose: To evaluate MLFF capability for molecule-surface interactions dominated by noncovalent forces [66].
Materials:
- Interface structure: 1,8-naphthyridine on graphene
- DFT reference: PBE-D3(BJ) with dense k-point sampling
- Training data: Various adsorption configurations
Procedure:
- Training Set Design:
- Sample multiple adsorption sites (top, bridge, hollow)
- Include various molecular orientations
- Incorporate vertical displacement from surface (2-5 Å)
Model Training:
- Use extended cutoffs (6-8 Å) for surface-molecule interactions
- Employ energy-weighting in loss function for binding configurations
- Validate with adsorption energy calculations
MD Simulations:
- Run 100-500 ps simulations of adsorbed molecule at 300 K
- Analyze diffusion pathways and residence times
Analysis:
- Compare binding energies with DFT reference
- Analyze angular orientation distributions
- Calculate diffusion coefficients
Validation Metrics: Binding energy error (< 50 meV), adsorption geometry, diffusion barriers, vibrational frequencies [66].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Computational Tools for MLFF Development and Validation
| Tool Category | Specific Solutions | Function | Application Context |
|---|---|---|---|
| QM Reference Codes | VASP, CP2K, Quantum ESPRESSO, PySCF, ORCA | Generate training data via DFT calculations [63] | Materials (periodic), molecules (molecular) systems [63] |
| MLFF Frameworks | MACE, NequIP, SO3krates, sGDML | Implement specific MLFF architectures | System-dependent selection [66] [62] [64] |
| Datasets | Alexandria, OC20, OMat24, QM-9, Materials Project | Provide training and benchmarking data [63] [67] | Model development and validation |
| MD Engines | LAMMPS, ASE, i-PI | Perform molecular dynamics simulations | Production MD with trained potentials |
| Analysis Tools | MDTraj, OVITO, pymatgen | Analyze trajectories and calculate observables | Validation and interpretation |
Implementation Guidelines for Material Validation Research
Architecture Selection Framework
Choose MLFF architectures based on specific research requirements:
- For data-efficient learning: NequIP demonstrates exceptional performance with limited training data, achieving accurate results with as few as 100 reference calculations [64].
- For exploration tasks: SO3krates provides optimal balance between discovering new conformations and maintaining simulation stability [62].
- For many-body interactions: MACE incorporates higher-order body-order terms for complex electronic interactions [66].
- For kernel-based approaches: sGDML and FCHL19* offer strong performance for small to medium-sized molecular systems [66].
Training Dataset Construction
The quality and representativeness of training datasets often outweigh architectural choices [66]. Implement active learning protocols to efficiently sample configuration space:
- Initial Sampling: Use normal mode sampling, MD at various temperatures, or random structural perturbations
- Uncertainty Quantification: Implement query-by-committee or dropout uncertainty estimates
- Iterative Refinement: Retrain models with strategically selected additional points
- Transfer Learning: Leverage pre-trained models on large datasets (Alexandria, OC20) with fine-tuning [67]
Special Considerations for Material Validation
- Long-range interactions: Implement physical priors or hybrid approaches for systems with significant electrostatic or dispersion forces [66]
- Phase transitions: Ensure training data includes intermediate configurations between phases
- Defect properties: Include defective configurations at appropriate concentrations
- Surface phenomena: Incorporate various surface terminations and adsorbate configurations
This comparative analysis demonstrates that modern MLFF architectures have reached a maturity level where multiple models can deliver reliable molecular dynamics simulations when properly trained and validated. The choice between MACE, SO3krates, NequIP, or other architectures should be guided by specific research constraints including available training data, computational resources, and target properties. For material validation research integrating DFT and machine learning, emphasis should be placed on developing complete, representative training datasets that adequately sample the relevant configuration space, with architectural selection playing a secondary role for many applications. Future development should focus on improving the description of long-range interactions and establishing standardized benchmarking protocols across diverse materials classes.
The integration of density functional theory (DFT) and machine learning (ML) is revolutionizing the predictive modeling of materials and biological systems in clinical and biomedical research. While DFT provides a first-principles quantum mechanical framework for calculating electronic structures, machine learning enhances this by correcting systematic errors, accelerating property predictions, and enabling high-throughput screening. This synergy is particularly critical in areas such as drug discovery, biomaterial development, and personalized medicine, where predictive accuracy directly impacts therapeutic outcomes and patient safety [5] [68] [69].
However, the translation of these computational approaches into clinically reliable tools faces significant challenges. Predictive models must overcome issues such as data scarcity, model interpretability, and validation against experimental benchmarks to be trusted in biomedical decision-making [68] [70]. This protocol details a structured framework for establishing trust in DFT-ML integrations, with a focus on rigorous validation, error correction, and practical implementation in biomedical contexts.
Table 1: Key Challenges in DFT-ML Integration for Biomedical Applications
| Challenge | Impact on Predictive Reliability | Potential Solution |
|---|---|---|
| DFT Energy Resolution Errors | Limited accuracy in formation enthalpy and phase stability predictions [5] | ML-based error correction using experimental benchmarks |
| Data Scarcity in Biological Systems | Poor model generalizability for complex biomolecules [68] | Transfer learning and data augmentation strategies |
| Model Interpretability | Limited adoption in clinical decision-making due to "black box" concerns [70] | Explainable AI (XAI) and feature importance analysis |
| Multi-Scale Validation | Disconnect between computational predictions and clinical outcomes [69] | Cross-platform benchmarking and experimental verification |
Foundational Concepts
Density Functional Theory in Biomedical Contexts
DFT serves as the computational foundation for understanding electronic structures in biological systems, from drug-target interactions to biomaterial properties. The Kohn-Sham formulation of DFT expresses the total energy of a system as a functional of the electronic charge density: [ E{\text{total}}[\rho] = T{\text{KS}}[\rho] + E{\text{XC}}[\rho] + E{\text{H}}[\rho] + E{\text{ext}}[\rho] ] where (T{\text{KS}}) represents the kinetic energy of non-interacting particles, (E{\text{XC}}) the exchange-correlation effects, (E{\text{H}}) the electrostatic interactions, and (E_{\text{ext}}) the external potential [68].
Despite its widespread use, DFT has inherent limitations in biomedical applications. Exchange-correlation functional approximations can introduce systematic errors in predicting reaction energetics and binding affinities, while self-interaction errors may lead to spurious charge transfer in molecular systems [68]. These limitations are particularly problematic when modeling transition metal complexes in enzymatic reactions or predicting protein-ligand binding energies, necessitating robust error correction methodologies.
Machine Learning Paradigms for Enhanced Accuracy
Machine learning approaches to improving DFT accuracy generally fall into three categories:
Machine-Learned Density Functionals: ML models trained to approximate exchange-correlation functionals using high-fidelity quantum chemistry or experimental data [68].
Hamiltonian Corrections: Structure-dependent models that correct fundamental errors in DFT approximations, such as self-interaction or delocalization errors [68].
Δ-ML Approaches: Post-DFT corrections that learn the difference between DFT predictions and accurate reference data [68].
For clinical and biomedical applications, Δ-ML methods have shown particular promise. For instance, neural networks trained to predict the discrepancy between DFT-calculated and experimentally measured formation enthalpies have demonstrated significant improvements in predictive accuracy for alloy systems relevant to biomedical implants [5].
Quantitative Assessment of Predictive Accuracy
Validating the performance of integrated DFT-ML approaches requires comprehensive quantitative assessment against experimental benchmarks. The following metrics provide a standardized framework for evaluating predictive accuracy across different biomedical applications.
Table 2: Performance Metrics for DFT-ML Models in Biomedical Predictions
| Application Domain | Key Predictive Task | Performance Metric | Reported Performance |
|---|---|---|---|
| MR-TADF Emitters for OLED Biosensing [69] | Photoluminescence Quantum Yield (PLQY) | Regression Accuracy (R²) | >0.85 (RF/LightGBM models) |
| Transition Metal Sulfides for Catalytic Therapy [71] | Band Gap Prediction | R² / RMSE | R²: 0.9989 (fine-tuned LLM) |
| Alloy Formation Enthalpy for Implants [5] | Enthalpy of Formation | Leave-One-Out Cross-Validation | Significant error reduction vs. pure DFT |
| Thermodynamic Stability [71] | Stability Classification | F1 Score | >0.775 (fine-tuned LLM) |
| Protein-Ligand Binding [70] | Binding Affinity Prediction | Mean Absolute Error | Varies by system (ML-enhanced) |
The exceptional performance of fine-tuned Large Language Models (LLMs) for predicting material properties highlights an emerging trend in computational biomedicine [71]. By processing textual descriptions of crystal structures directly, these models eliminate complex feature engineering while maintaining high accuracy, even with limited labeled data (e.g., 554 compounds in the transition metal sulfide study).
Experimental Protocols
Protocol 1: ML-Corrected DFT for Biomaterial Property Prediction
This protocol outlines a standardized workflow for improving the accuracy of DFT-predicted biomaterial properties using machine learning correction, adapted from successful applications in alloy thermodynamics [5].
Workflow Visualization
Step-by-Step Procedure
System Definition and Data Curation
- Define the biomaterial system composition and structure space
- Perform high-throughput DFT calculations using consistent parameters (e.g., PBE functional, k-point mesh 17×17×17 for cubic systems) [5]
- Collect experimental reference data from reliable sources, filtering out uncertain measurements
- For a study on Ni-Al-Pd alloys: 80+ compounds with formation enthalpies from experimental literature [5]
Feature Engineering and Selection
Model Architecture and Training
- Implement a Multi-Layer Perceptron (MLP) with 3 hidden layers [5]
- Apply Leave-One-Out Cross-Validation (LOOCV) and k-fold cross-validation
- Optimize hyperparameters (layer sizes, activation functions, learning rate)
- Train to minimize the difference between DFT predictions and experimental values
Validation and Performance Assessment
- Calculate key metrics: R², RMSE, MAE against test set
- Apply domain-specific validation (e.g., phase boundary accuracy for alloys)
- Compare against baseline DFT-only predictions
Protocol 2: Fine-Tuned LLMs for Biomaterial Property Prediction
This protocol adapts the emerging approach of using fine-tuned Large Language Models for predicting material properties, demonstrating exceptional performance with limited data [71].
Workflow Visualization
Step-by-Step Procedure
Data Acquisition and Preprocessing
- Extract transition metal sulfide data from Materials Project API using specific criteria [71]:
- Formation energy < 500 eV/atom
- Energy above hull < 150 eV/atom
- Transition metals (Sc-Zn, Y-Cd, La-Hg) combined with sulfur
- Apply quality filters: remove unconverged relaxations (forces > 0.05 eV/Å), disordered structures, inconsistent calculations
- From 729 initial compounds, select 554 high-quality samples for training [71]
- Extract transition metal sulfide data from Materials Project API using specific criteria [71]:
Textual Description Generation
- Process crystal structures through robocrystallographer to generate standardized textual descriptions [71]
- Create natural language narratives capturing atomic arrangements, bond properties, and electronic characteristics
- Format training data as JSONL with structure-property pairs
Iterative Model Fine-Tuning
- Initialize with GPT-3.5-turbo or domain-specific foundation model
- Conduct multiple fine-tuning iterations (9+ cycles demonstrated) [71]
- Implement structured prompt templates for consistency
- Track loss convergence and target high-loss data points for retraining
Validation and Benchmarking
- Compare against traditional ML (Random Forest, SVM, XGBoost) and GNN baselines
- Evaluate band gap prediction (R², RMSE) and stability classification (F1 score)
- Assess generalization across diverse material structures
Protocol 3: DFT-Enhanced ML for Photoluminescence Efficiency Prediction
This protocol details a unified DFT-ML framework for predicting photoluminescence quantum yield (PLQY) of molecular emitters, with applications in biosensing and diagnostic imaging [69].
Workflow Visualization
Step-by-Step Procedure
Standardized Dataset Construction
Key Descriptor Identification
- Identify emission transition dipole moment (TDM) as the most influential PLQY descriptor [69]
- Develop fast, fingerprint-based TDM prediction model for high-throughput screening
- Uncover substructural motifs governing TDM through model interpretation
Model Training and Validation
- Implement ensemble methods (Random Forest, LightGBM, XGBoost) using sklearn-ensemble [69]
- Split training and test sets via sklearn module
- Validate predictions against experimental PLQY measurements
- Achieve high correlation between predicted and experimental values
Inverse Design and Experimental Verification
Successful implementation of DFT-ML protocols requires specialized computational tools and resources. The following table summarizes key components for establishing a robust research workflow.
Table 3: Essential Research Reagents and Computational Resources
| Resource Category | Specific Tools/Platforms | Function in DFT-ML Workflow |
|---|---|---|
| DFT Calculation Software | EMTO-CPA [5], Gaussian 16 [69] | First-principles electronic structure calculations for training data generation |
| Machine Learning Frameworks | sklearn-ensemble, XGBoost, LightGBM [69] | Implementation of ML correction models and property predictors |
| Large Language Models | GPT-3.5-turbo, GPT-4.0 [71] | Fine-tuned property prediction from textual material descriptions |
| Material Databases | Materials Project API [71], Alexandria database [72] | Source of curated structural and property data for training |
| Descriptor Generation | Robocrystallographer [71] | Automated generation of textual crystal structure descriptions |
| Validation Tools | MatBench [72], Cross-validation modules | Performance assessment and model benchmarking |
Concluding Remarks
The integration of DFT and machine learning represents a paradigm shift in predictive materials modeling for clinical and biomedical applications. The protocols outlined in this document provide a structured framework for achieving and validating predictive accuracy in these critical domains. Key insights from successful implementations include:
Hybrid Approaches Outperform Single Methods: ML-corrected DFT consistently demonstrates superior accuracy compared to pure DFT or standalone ML predictions, particularly for complex thermodynamic properties [5].
Data Quality Over Quantity: Carefully curated datasets of a few hundred compounds can yield exceptional predictive accuracy when processed through appropriate models, as demonstrated by fine-tuned LLMs achieving R² > 0.99 with only 554 training samples [71].
Multi-Scale Validation is Essential: Trust in predictive models requires validation across computational benchmarks, experimental measurements, and ultimately clinical relevance [69] [70].
As these methodologies continue to evolve, their implementation in biomedical contexts will increasingly rely on standardized protocols, rigorous validation, and transparent reporting of limitations. The establishment of trust in these predictive tools will ultimately enable their translation from computational research to clinical impact, accelerating the development of novel therapies, diagnostic agents, and biomedical materials.
Conclusion
The integration of DFT and machine learning marks a paradigm shift in computational materials validation, successfully bridging the gap between quantum-mechanical accuracy and high-throughput screening. The foundational principles establish a solid framework, while advanced methodologies like specialized MLFFs and modular frameworks provide powerful, actionable tools. Overcoming data and generalization challenges through domain adaptation and rigorous benchmarking is essential for building trustworthy models. As these hybrid methods mature, their implications for biomedical and clinical research are profound, promising to drastically accelerate the design of novel drug delivery systems, biomaterials, and therapeutic agents. Future progress hinges on developing more data-efficient learning schemes, improving model interpretability, and establishing standardized validation protocols tailored to the specific reliability requirements of drug development, ultimately paving the way for a new era of AI-driven material discovery.
References
- 1. Predicting electronic structures at any length scale with ... [https://www.nature.com/articles/s41524-023-01070-z]
- 2. Efficient and Scalable Density Functional Theory ... [https://arxiv.org/html/2502.01171v2]
- 3. A deep learning framework to emulate density functional ... [https://www.nature.com/articles/s41524-023-01115-3]
- 4. Materials Learning Algorithms (MALA): Scalable machine ... [https://www.sciencedirect.com/science/article/pii/S0010465525001560]
- 5. Machine learning for improved density functional theory ... [https://www.nature.com/articles/s41598-025-02088-7]
- 6. Integrating Density Functional Theory with Deep Neural ... [https://arxiv.org/html/2503.13067v3]
- 7. A Record-Breaking Dataset to Train AI Models has Launched [https://newscenter.lbl.gov/2025/05/14/computational-chemistry-unlocked-a-record-breaking-dataset-to-train-ai-models-has-launched/]
- 8. [2505.08762] The Open Molecules 2025 (OMol25) Dataset, ... [https://arxiv.org/abs/2505.08762]
- 9. AI/ML as a Security Team Force Multiplier [https://www.cybereason.com/blog/ai/ml-as-a-security-team-force-multiplier]
- 10. The Force Multiplier: A Playbook for Training a Traditional ... [https://www.linkedin.com/pulse/force-multiplier-playbook-training-traditional-process-anton-dubov-nkwnc]
- 11. Towards Universal Material Property Prediction with Deep ... [https://arxiv.org/html/2510.13207v1]
- 12. MD-HIT: Machine learning for material property prediction ... [https://www.nature.com/articles/s41524-024-01426-z]
- 13. Integrating density functional theory with machine learning ... [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d4cp03397c]
- 14. Applications of density functional theory and machine ... [https://www.sciencedirect.com/science/article/pii/S2949822825002011]
- 15. Machine learning-enabled optoelectronic material discovery [https://www.oaepublish.com/articles/jmi.2025.13]
- 16. Materials-discovery workflow guided by symbolic ... [https://www.nature.com/articles/s41524-025-01596-4]
- 17. Scaling deep learning for materials discovery [https://www.nature.com/articles/s41586-023-06735-9]
- 18. MatFold: systematic insights into materials discovery models... [https://pubs.rsc.org/en/content/articlelanding/2025/dd/d4dd00250d]
- 19. Machine Learning-Driven Materials Discovery [https://www.sciencedirect.com/science/article/abs/pii/S235221432500139X]
- 20. a review of machine learning approaches to cathode material ... [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d5ta04723d?page=search]
- 21. The mastery of details in the workflow of materials machine ... [https://www.nature.com/articles/s41524-024-01331-5]
- 22. Accurate, interpretable predictions of materials properties ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10591138/]
- 23. Experimental validation, anyone? [https://www.nature.com/articles/s43588-023-00462-x]
- 24. Nanoscale reference and test materials for the validation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12003566/]
- 25. : A tool for constructing accurate DPmoire ... machine learning force [https://arxiv.org/html/2412.19333v1]
- 26. JiaxuanLiu-Arsko/ DPmoire : A tool for constructing accurate machine ... [https://github.com/JiaxuanLiu-Arsko/DPmoire]
- 27. Error analysis and hyperparameter optimization of machine ... [https://www.vasp.at/tutorials/latest/mlff/part1/]
- 28. Best practices for machine-learned force fields [https://www.vasp.at/wiki/index.php/Best_practices_for_machine-learned_force_fields]
- 29. From density to functional ... theory machine learning predictive [https://pmc.ncbi.nlm.nih.gov/articles/PMC11637019/]
- 30. (PDF) Machine Methods to Learning ... Predict Density Functional [https://www.academia.edu/33502697/Machine_Learning_Methods_to_Predict_Density_Functional_Theory_B3LYP_Energies_of_HOMO_and_LUMO_Orbitals]
- 31. A DFT study to evaluate the modulation in the band , gap , and... elastic [https://pubs.rsc.org/en/content/articlelanding/2025/ma/d5ma00413f]
- 32. Large language models for material property predictions : elastic ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00061k]
- 33. -driven Machine of learning characteristics in... prediction band gap [https://link.springer.com/article/10.1007/s00542-025-05902-4]
- 34. for improved Machine ... learning density functional theory [https://www.nature.com/articles/s41598-025-02088-7?error=cookies_not_supported&code=6c0634dd-ca66-4478-a996-2bdd0ff3b01a]
- 35. Reinforce crystal material property prediction with ... [https://www.sciencedirect.com/science/article/abs/pii/S0927025624001794]
- 36. Crystalline Material Discovery in the Age of Artificial ... [https://arxiv.org/html/2408.08044v4]
- 37. AI trends in 2025: Graph Neural Networks [https://assemblyai.com/blog/ai-trends-graph-neural-networks]
- 38. AI for Crystal Materials: models and benchmarks [https://github.com/WanyuGroup/AI-for-Crystal-Materials]
- 39. : A Modular Deep Learning MoMa for Material Property... Framework [https://arxiv.org/html/2502.15483v2]
- 40. Transfer Learning vs Domain Adaptation [https://www.baeldung.com/cs/transfer-learning-vs-domain-adaptation]
- 41. Transfer Learning In Domain Adaptation [https://www.meegle.com/en_us/topics/transfer-learning/transfer-learning-in-domain-adaptation]
- 42. Active discovery of organic semiconductors [https://www.nature.com/articles/s41467-021-22611-4]
- 43. Machine learning assisted designing of organic ... [https://www.sciencedirect.com/science/article/abs/pii/S1387700323002228]
- 44. Machine learning for perovskite materials design and ... [https://www.nature.com/articles/s41524-021-00495-8]
- 45. Probing out-of-distribution generalization in machine ... [https://www.nature.com/articles/s43246-024-00731-w]
- 46. The JARVIS Infrastructure is All You Need for Materials ... [https://arxiv.org/html/2503.04133v1]
- 47. EMFF-2025: a general neural network potential for ... [https://www.nature.com/articles/s41524-025-01809-w]
- 48. Stability-Aware Training of Machine Learning Force Fields ... [https://arxiv.org/html/2402.13984v3]
- 49. Improving robustness and training efficiency of machine ... [https://arxiv.org/html/2504.15925v1]
- 50. Improving robustness and training efficiency of machine ... [https://www.alphaxiv.org/overview/2504.15925v1]
- 51. Advancements in Material Using Machine Learning Property Prediction [https://scisimple.com/en/articles/2025-07-20-advancements-in-material-property-prediction-using-machine-learning--a9pg0gz]
- 52. A Comprehensive Guide to AI-Ready Data Management in ... [https://cloudsoda.io/mastering-storage-optimization-a-comprehensive-guide-to-ai-ready-data-management-in-2025/]
- 53. Frontiers | Enhancing phase change thermal energy storage material ... [https://www.frontiersin.org/journals/materials/articles/10.3389/fmats.2025.1616233/full]
- 54. A combined ML and DFT strategy for the prediction of dye candidates... [https://www.nature.com/articles/s41524-025-01521-9?error=cookies_not_supported]
- 55. Crash testing machine learning force fields for molecules ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc06529h]
- 56. The ML FMEA: An Introduction - Torc ... [https://torc.ai/knowledge-center/publications/the-ml-fmea-an-introduction/]
- 57. Crash testing machine learning force fields for molecules ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc06530a]
- 58. Harnessing DFT and machine learning for accurate optical ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d4nr03702b]
- 59. Machine learning-driven material intelligence research and ... [https://www.sciopen.com/article/10.26599/NR.2025.94908095]
- 60. Validating Molecular Dynamics Simulations Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420231/]
- 61. Testing for physical validity in molecular simulations | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0202764]
- 62. A Euclidean transformer for fast and stable machine ... [https://www.nature.com/articles/s41467-024-50620-6]
- 63. An Introduction to Neural Network Potentials - Rowan Scientific [https://rowansci.com/publications/introduction-to-nnps]
- 64. E(3)-equivariant graph neural networks for data-efficient ... [https://www.nature.com/articles/s41467-022-29939-5]
- 65. Theoretical background of NequIP - Zhengda notes! [https://hezhengda.me/en/latest/ML_potential/NequIP/NequIP_theory.html]
- 66. Crash testing machine learning force fields for molecules ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc06530a]
- 67. Improving machine-learning models in materials science ... [https://www.sciencedirect.com/science/article/pii/S2542529324002360]
- 68. for Machine in learning approximations accuracy density functional [https://arxiv.org/html/2311.00196v2]
- 69. DFT-enhanced machine learning for accurate PLQY ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725109893]
- 70. Transformative Role of Artificial Intelligence in Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12406033/]
- 71. Fine-Tuned Large Language Models for High-Accuracy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12387436/]
- 72. Universal Machine Learning Potentials under Pressure [https://arxiv.org/html/2508.17792v1]